当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Study of Iron Porphyrin Nitrene: Formation Mechanism, Electronic Nature, and Intermolecular C-H Amination.
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.inorgchem.9b02216 Xinyi Li 1 , Lihua Dong 2 , Yongjun Liu 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.inorgchem.9b02216 Xinyi Li 1 , Lihua Dong 2 , Yongjun Liu 1
Affiliation
|
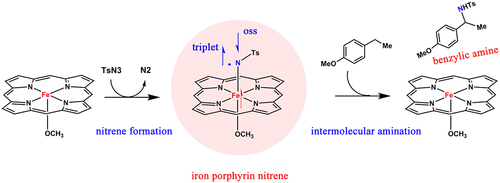
The formation mechanism and electronic structures of iron porphyrin nitrene intermediates, as well as the nitrene-mediated intermolecular C-H amination, have been studied by performing DFT and ab initio complete active space self-consistent field (CASSCF) calculations. Compared with that of cobalt porphyrin nitrene and iron porphyrin carbene, the formation of iron porphyrin nitrene shows similar but different characteristics. The common feature is that all their formation is required to undergo the "far" or "close" complexes, but these complexes correspond to different energies relative to their respective reactants (isolated metalloporphyrins and azides), which is considered as one main reason to determine the reaction barriers. The overall free energy barrier for the formation of iron porphyrin nitrene was calculated to be 10.6 kcal/mol on a triplet-state surface, which is lower than those of cobalt porphyrin nitrene and iron porphyrin carbene. The departure of N2 from the close complexes formed by iron porphyrin and tosyl azide is nearly barrierless. For iron porphyrin nitrene, both CASSCF and unrestricted DFT calculations revealed that the triplet and open-shell singlet complexes correspond to very similar energies. The triplet nitrene complex can be described as [(por)(-OCH3)FeII═NTs]- ↔ [(por)(-OCH3)FeIII═N•-Ts]- ↔ [(por)(-OCH3)FeIV═N2-Ts]-, while the oss nitrene complex can be described as [(por)(-OCH3)FeIII-N•-Ts]-. Since the N atom bears a similar spin density as in cobalt porphyrin nitrene, the iron porphyrin nitrene exhibits similar activity in hydrogen abstraction. In addition, the intermolecular C-H amination catalyzed by iron porphyrin nitrene follows the hydrogen atom abstraction/radical recombination mechanism with a free energy barrier of 7.1 kcal/mol on the triplet-state surface. In general, the medium reactivity and easily prepared characteristic of iron porphyrin nitrene makes it a potential catalyst for C-H amination.
中文翻译:

铁卟啉镍的理论研究:形成机理,电子性质和分子间CH胺化。
通过进行DFT和从头算起的完整活性空间自洽场(CASSCF)计算,研究了卟啉铁氮中间体的形成机理和电子结构,以及氮介导的分子间CH胺化。与钴卟啉氮和卟啉铁卡宾相比,卟啉氮的形成具有相似但不同的特征。共同特征是要求它们的所有形成都经历“远”或“近”配合物,但是这些配合物相对于其各自的反应物(分离的金属卟啉和叠氮化物)对应于不同的能量,这被认为是确定反应障碍。形成铁卟啉氮的总自由能垒经计算为10。在三重态表面上为6 kcal / mol,低于钴卟啉氮和铁卟啉卡宾。N2与由卟啉铁和甲苯磺酰基叠氮化物形成的紧密络合物的离开几乎是无障碍的。对于卟啉亚铁,CASSCF和无限制DFT计算均显示三重态和开壳单重态络合物对应于非常相似的能量。三重态氮络合物可描述为[[por](-OCH3)FeII═NTs]-↔[(por)(-OCH3)FeIII═N•-Ts]-↔[(por)(-OCH3)FeIV═N2 -Ts]-,而oss氮烯络合物可描述为[(por)(-OCH3)FeIII-N•-Ts]-。由于N原子具有与钴卟啉氮类似的自旋密度,因此卟啉铁氮在夺氢方面具有相似的活性。此外,卟啉亚铁催化的分子间CH胺化反应遵循氢原子提取/自由基复合机理,在三重态表面上的自由能垒为7.1 kcal / mol。通常,卟啉亚铁的介质反应性和易于制备的特性使其成为潜在的CH胺化催化剂。
更新日期:2020-01-13
中文翻译:
铁卟啉镍的理论研究:形成机理,电子性质和分子间CH胺化。
通过进行DFT和从头算起的完整活性空间自洽场(CASSCF)计算,研究了卟啉铁氮中间体的形成机理和电子结构,以及氮介导的分子间CH胺化。与钴卟啉氮和卟啉铁卡宾相比,卟啉氮的形成具有相似但不同的特征。共同特征是要求它们的所有形成都经历“远”或“近”配合物,但是这些配合物相对于其各自的反应物(分离的金属卟啉和叠氮化物)对应于不同的能量,这被认为是确定反应障碍。形成铁卟啉氮的总自由能垒经计算为10。在三重态表面上为6 kcal / mol,低于钴卟啉氮和铁卟啉卡宾。N2与由卟啉铁和甲苯磺酰基叠氮化物形成的紧密络合物的离开几乎是无障碍的。对于卟啉亚铁,CASSCF和无限制DFT计算均显示三重态和开壳单重态络合物对应于非常相似的能量。三重态氮络合物可描述为[[por](-OCH3)FeII═NTs]-↔[(por)(-OCH3)FeIII═N•-Ts]-↔[(por)(-OCH3)FeIV═N2 -Ts]-,而oss氮烯络合物可描述为[(por)(-OCH3)FeIII-N•-Ts]-。由于N原子具有与钴卟啉氮类似的自旋密度,因此卟啉铁氮在夺氢方面具有相似的活性。此外,卟啉亚铁催化的分子间CH胺化反应遵循氢原子提取/自由基复合机理,在三重态表面上的自由能垒为7.1 kcal / mol。通常,卟啉亚铁的介质反应性和易于制备的特性使其成为潜在的CH胺化催化剂。









































 京公网安备 11010802027423号
京公网安备 11010802027423号