Solid State Ionics ( IF 3.0 ) Pub Date : 2020-01-10 , DOI: 10.1016/j.ssi.2019.115192 David Case , Adam J. McSloy , Ryan Sharpe , Stephen R. Yeandel , Thomas Bartlett , James Cookson , Enkhtsetseg Dashjav , Frank Tietz , C.M. Naveen Kumar , Pooja Goddard
|
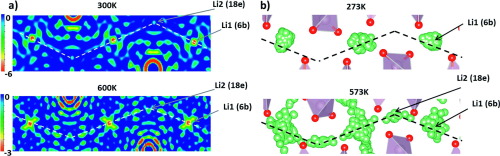
New solid state electrolytes are becoming increasingly sought after in the drive to replace flammable liquid electrolytes. To this end, several Li conducting solids have been identified as promising candidates including Li stuffed garnets and more recently Li-rich materials such as Li1+xAlxTi2−x(PO4)3 with 0.3< x <0.5. However, the structure/property relationships of LATP are incredibly sensitive to synthesis conditions and therefore challenging to optimise. In this joint computational and experimental investigation, we examine the structural sensitivities by modelling the site occupancies at varying temperature, which clarifies previously reported discrepancies of the crystal structures. Furthermore, we investigate the Li ion transport properties which have not reported computationally before. We confirm from our simulations that the migration pathway only involves the M1(6b) and M2(18e) site, in excellent agreement with the neutron diffraction data, clarifying all past controversies regarding the Li ion occupancies in LATP. Interestingly, we calculate low migration barriers (0.3 eV) in line with experimental findings but also show evidence of Li ion trapping on Al doping in LATP (where x = 0.4), possibly explaining the experimental observation that the Li ion conductivity does not improve above x = 0.3, due to a stronger repulsion between Li+–>Ti4+ compared to Li+–>Al3+. Furthermore, our calculated ionic conductivities are in excellent agreement with experimental values, highlighting the robustness of our computational models.
中文翻译:
0.3 < x <0.5的富锂Li 1+ x Al x Ti 2− x(PO 4)3的结构和离子迁移:计算与实验研究相结合
在替代可燃液体电解质的驱动中,新型固态电解质正变得越来越受欢迎。为此,已经确定了几种锂导电固体是有前途的候选材料,包括锂填充石榴石和最近的富含锂的材料,例如0.3 < x的Li 1+ x Al x Ti 2− x(PO 4)3<0.5。但是,LATP的结构/性质关系对合成条件极为敏感,因此难以优化。在这项联合的计算和实验研究中,我们通过对不同温度下的位点占用进行建模来检查结构敏感性,从而澄清了先前报道的晶体结构差异。此外,我们研究了锂离子的输运性质,这在以前没有进行过计算。我们从模拟中证实,迁移路径仅涉及M1(6b)和M2(18e)位置,与中子衍射数据非常吻合,从而澄清了有关LATP中锂离子占用的所有以往争议。有趣的是,我们计算出较低的迁移障碍(0。+ –> Ti 4+与Li + –> Al 3+相比。此外,我们计算出的离子电导率与实验值非常吻合,突出了我们计算模型的鲁棒性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号