当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reconciling Simulated Ensembles of Apomyoglobin with Experimental Hydrogen/Deuterium Exchange Data Using Bayesian Inference and Multiensemble Markov State Models.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-23 , DOI: 10.1021/acs.jctc.9b01240 Hongbin Wan 1 , Yunhui Ge 1 , Asghar Razavi 1 , Vincent A Voelz 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-23 , DOI: 10.1021/acs.jctc.9b01240 Hongbin Wan 1 , Yunhui Ge 1 , Asghar Razavi 1 , Vincent A Voelz 1
Affiliation
|
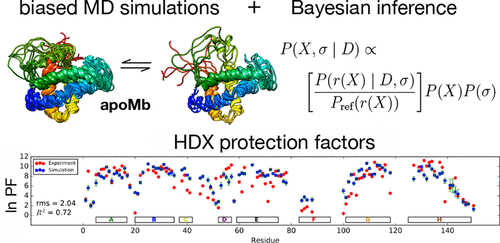
Hydrogen/deuterium exchange (HDX) is a powerful technique to investigate protein conformational dynamics at amino acid resolution. Because HDX provides a measurement of solvent exposure of backbone hydrogens, ensemble-averaged over potentially slow kinetic processes, it has been challenging to use HDX protection factors to refine structural ensembles obtained from molecular dynamics simulations. This entails dual challenges: (1) identifying structural observables that best correlate with backbone amide protection from exchange and (2) restraining these observables in molecular simulations to model ensembles consistent with experimental measurements. Here, we make significant progress on both fronts. First, we describe an improved predictor of HDX protection factors from structural observables in simulated ensembles, parametrized from ultralong molecular dynamics simulation trajectory data, with a Bayesian inference approach used to retain the full posterior distribution of model parameters. We next present a new method for obtaining simulated ensembles in agreement with experimental HDX protection factors, in which molecular simulations are performed at various temperatures and restraint biases and used to construct multiensemble Markov State Models (MSMs). Finally, the BICePs (Bayesian Inference of Conformational Populations) algorithm is then used with our HDX protection factor predictor to infer which thermodynamic ensemble agrees best with the experiment and estimate populations of each conformational state in the MSM. To illustrate the approach, we use a combination of HDX protection factor restraints and chemical shift restraints to model the conformational ensemble of apomyoglobin at pH 6. The resulting ensemble agrees well with the experiment and gives insight into the all-atom structure of disordered helices F and H in the absence of heme.
中文翻译:

使用贝叶斯推断和多集合马尔可夫状态模型将模拟的肌球蛋白与实验氢/氘交换数据协调一致。
氢/氘交换(HDX)是一项功能强大的技术,可在氨基酸分辨率下研究蛋白质构象动力学。由于HDX可以测量骨架氢的溶剂暴露量(在潜在的缓慢动力学过程中进行平均),因此使用HDX保护因子来完善从分子动力学模拟获得的结构体一直是一项挑战。这带来了双重挑战:(1)确定与骨架酰胺保护免受交换最相关的结构可观察物,(2)在分子模拟中限制这些可观察物以模拟与实验测量结果一致的集合体。在这里,我们在这两个方面都取得了重大进展。首先,我们从模拟合奏中的结构观测值描述了HDX保护因子的改进预测因子,从超长分子动力学模拟轨迹数据中参数化,并使用贝叶斯推理方法来保留模型参数的全部后验分布。接下来,我们提出一种与实验HDX保护因子一致的获得模拟合奏的新方法,该方法在各种温度和约束偏差下进行分子模拟,并用于构建多集合马尔可夫状态模型(MSM)。最后,然后将BICePs(构象种群的贝叶斯推断)算法与我们的HDX保护因子预测器一起使用,以推断哪种热力学集合与实验最吻合,并估计MSM中每个构象状态的种群。为了说明这种方法,
更新日期:2020-01-24
中文翻译:
使用贝叶斯推断和多集合马尔可夫状态模型将模拟的肌球蛋白与实验氢/氘交换数据协调一致。
氢/氘交换(HDX)是一项功能强大的技术,可在氨基酸分辨率下研究蛋白质构象动力学。由于HDX可以测量骨架氢的溶剂暴露量(在潜在的缓慢动力学过程中进行平均),因此使用HDX保护因子来完善从分子动力学模拟获得的结构体一直是一项挑战。这带来了双重挑战:(1)确定与骨架酰胺保护免受交换最相关的结构可观察物,(2)在分子模拟中限制这些可观察物以模拟与实验测量结果一致的集合体。在这里,我们在这两个方面都取得了重大进展。首先,我们从模拟合奏中的结构观测值描述了HDX保护因子的改进预测因子,从超长分子动力学模拟轨迹数据中参数化,并使用贝叶斯推理方法来保留模型参数的全部后验分布。接下来,我们提出一种与实验HDX保护因子一致的获得模拟合奏的新方法,该方法在各种温度和约束偏差下进行分子模拟,并用于构建多集合马尔可夫状态模型(MSM)。最后,然后将BICePs(构象种群的贝叶斯推断)算法与我们的HDX保护因子预测器一起使用,以推断哪种热力学集合与实验最吻合,并估计MSM中每个构象状态的种群。为了说明这种方法,








































 京公网安备 11010802027423号
京公网安备 11010802027423号