当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
uMBD: A Materials-Ready Dispersion Correction that Uniformly Treats Metallic, Ionic, and van der Waals Bonding
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-01-10 , DOI: 10.1021/jacs.9b11589 Minho Kim 1, 2 , Won June Kim 3 , Timothy Gould 4 , Eok Kyun Lee 1 , Sébastien Lebègue 2 , Hyungjun Kim 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2020-01-10 , DOI: 10.1021/jacs.9b11589 Minho Kim 1, 2 , Won June Kim 3 , Timothy Gould 4 , Eok Kyun Lee 1 , Sébastien Lebègue 2 , Hyungjun Kim 1
Affiliation
|
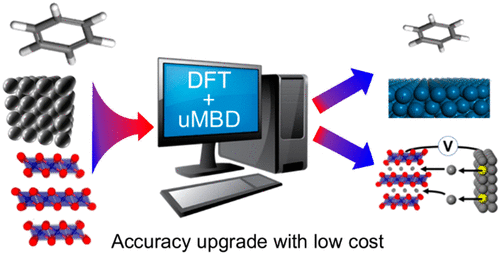
Materials design increasingly relies on first-principles calculations for screening important candidates and for under-standing quantum mechanisms. Density functional theory (DFT) is by far the most popular first-principles approach due to its efficiency and accuracy. However, to accurately predict structures and thermodynamics, DFT must be paired with a van der Waals (vdW) dispersion correction. Therefore, such corrections have been the subject of intense scrutiny in re-cent years. Despite significant successes in organic molecules, no existing model can adequately cover the full range of common materials, from metals to ionic solids, hampering the applications of DFT for modern problems such as battery design. Here, we introduce a universally optimized vdW-corrected DFT method that demonstrates an unbiased reliability for predicting molecular, layered, ionic, metallic, and hybrid materials without incurring a large computational overhead. We use our method to accurately predict the intercalation potentials of layered electrode materials of a Li-ion battery system - a problem for which the existing state-of-the-art methods fail. Thus, we envisage broad use of our method in the design of chemo-physical processes of new materials.
中文翻译:

uMBD:一种可统一处理金属、离子和范德华键合的材料就绪色散校正
材料设计越来越依赖于第一性原理计算来筛选重要的候选物和理解量子机制。密度泛函理论 (DFT) 因其效率和准确性而成为迄今为止最受欢迎的第一性原理方法。然而,为了准确预测结构和热力学,DFT 必须与范德华 (vdW) 色散校正配对。因此,此类更正近年来一直受到严格审查。尽管在有机分子方面取得了重大成功,但现有模型无法充分涵盖从金属到离子固体的所有常见材料,这阻碍了 DFT 在电池设计等现代问题中的应用。在这里,我们介绍了一种普遍优化的 vdW 校正 DFT 方法,该方法展示了预测分子的无偏可靠性,分层、离子、金属和混合材料,而不会产生大量计算开销。我们使用我们的方法来准确预测锂离子电池系统分层电极材料的嵌入电位 - 这是现有最先进方法无法解决的问题。因此,我们设想在新材料的化学物理过程设计中广泛使用我们的方法。
更新日期:2020-01-10
中文翻译:
uMBD:一种可统一处理金属、离子和范德华键合的材料就绪色散校正
材料设计越来越依赖于第一性原理计算来筛选重要的候选物和理解量子机制。密度泛函理论 (DFT) 因其效率和准确性而成为迄今为止最受欢迎的第一性原理方法。然而,为了准确预测结构和热力学,DFT 必须与范德华 (vdW) 色散校正配对。因此,此类更正近年来一直受到严格审查。尽管在有机分子方面取得了重大成功,但现有模型无法充分涵盖从金属到离子固体的所有常见材料,这阻碍了 DFT 在电池设计等现代问题中的应用。在这里,我们介绍了一种普遍优化的 vdW 校正 DFT 方法,该方法展示了预测分子的无偏可靠性,分层、离子、金属和混合材料,而不会产生大量计算开销。我们使用我们的方法来准确预测锂离子电池系统分层电极材料的嵌入电位 - 这是现有最先进方法无法解决的问题。因此,我们设想在新材料的化学物理过程设计中广泛使用我们的方法。








































 京公网安备 11010802027423号
京公网安备 11010802027423号