当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ligand binding free energy and kinetics calculation in 2020
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2020-01-09 , DOI: 10.1002/wcms.1455 Vittorio Limongelli 1, 2
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2020-01-09 , DOI: 10.1002/wcms.1455 Vittorio Limongelli 1, 2
Affiliation
|
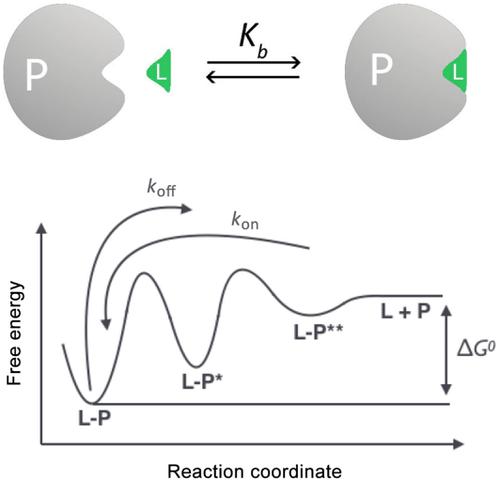
Ligand/protein binding (LPB) is a major topic in medicine, chemistry and biology. Since the advent of computers, many scientists have put efforts in developing theoretical models that could decode the alphabet of the LPB interaction. The success of this task passes by the resolution of the molecular mechanism of LPB. In the past century, major attention was dedicated to the thermodynamics of LPB, while more recent studies have revealed that ligand (un)binding kinetics is at least as important as ligand binding thermodynamics in determining the drug in vivo efficacy. In the present review, we introduce the most widely used computational methods to study LPB thermodynamics and kinetics. It is important to say that no method outperforms another, they all have pros and cons and the choice of the user should take carefully into account the system under investigation, the available structural and experimental data, and the goal of the research. A perspective on future directions of method development and research on LPB concludes the discussion.
中文翻译:

2020年配体结合自由能和动力学计算
配体/蛋白质结合(LPB)是医学,化学和生物学领域的主要话题。自从计算机出现以来,许多科学家就致力于开发可以解码LPB交互字母的理论模型。该任务的成功通过了LPB分子机制的解析。在过去的一个世纪中,主要的注意力集中在了LPB的热力学上,而最近的研究表明,在确定药物体内功效时,配体(非)结合动力学至少与配体结合热力学同等重要。在当前的审查中,我们介绍最广泛使用的计算方法来研究LPB热力学和动力学。重要的是要说没有一种方法能胜过另一种方法,它们各有利弊,用户的选择应仔细考虑所研究的系统,可用的结构和实验数据以及研究目标。对LPB方法开发和研究的未来方向的展望总结了讨论。
更新日期:2020-01-09
中文翻译:
2020年配体结合自由能和动力学计算
配体/蛋白质结合(LPB)是医学,化学和生物学领域的主要话题。自从计算机出现以来,许多科学家就致力于开发可以解码LPB交互字母的理论模型。该任务的成功通过了LPB分子机制的解析。在过去的一个世纪中,主要的注意力集中在了LPB的热力学上,而最近的研究表明,在确定药物体内功效时,配体(非)结合动力学至少与配体结合热力学同等重要。在当前的审查中,我们介绍最广泛使用的计算方法来研究LPB热力学和动力学。重要的是要说没有一种方法能胜过另一种方法,它们各有利弊,用户的选择应仔细考虑所研究的系统,可用的结构和实验数据以及研究目标。对LPB方法开发和研究的未来方向的展望总结了讨论。










































 京公网安备 11010802027423号
京公网安备 11010802027423号