当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-07 , DOI: 10.1021/acs.jcim.9b00843 Lucia Fusani 1, 2 , David S Palmer 2 , Don O Somers 3 , Ian D Wall 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-07 , DOI: 10.1021/acs.jcim.9b00843 Lucia Fusani 1, 2 , David S Palmer 2 , Don O Somers 3 , Ian D Wall 1
Affiliation
|
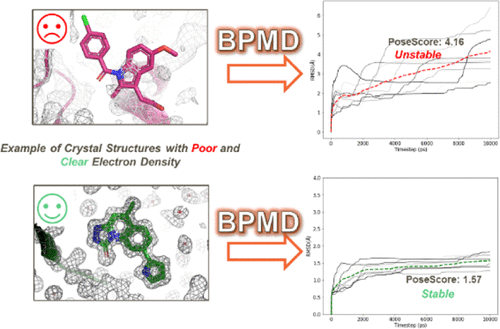
Identification of correct protein-ligand binding poses is important in structure-based drug design and crucial for the evaluation of protein-ligand binding affinity. Protein-ligand coordinates are commonly obtained from crystallography experiments that provide a static model of an ensemble of conformations. Binding pose metadynamics (BPMD) is an enhanced sampling method that allows for an efficient assessment of ligand stability in solution. Ligand poses that are unstable under the bias of the metadynamics simulation are expected to be infrequently occupied in the energy landscape, thus making minimal contributions to the binding affinity. Here, the robustness of the method is studied using crystal structures with ligands known to be incorrectly modeled, as well as 63 structurally diverse crystal structures with ligand fit to electron density from the Twilight database. Results show that BPMD can successfully differentiate compounds whose binding pose is not supported by the electron density from those with well-defined electron density.
中文翻译:

使用结合姿势代谢动力学研究蛋白质晶体结构中的配体稳定性。
正确的蛋白质-配体结合姿势的鉴定在基于结构的药物设计中很重要,对于评估蛋白质-配体结合亲和力至关重要。蛋白质-配体坐标通常从晶体学实验中获得,该晶体学实验提供了构象整体的静态模型。结合姿势元动力学(BPMD)是一种增强的采样方法,可以有效评估溶液中的配体稳定性。在元动力学模拟的偏压下不稳定的配体姿势预计将很少出现在能量图中,因此对结合亲和力的贡献很小。在此,使用已知配体模型错误的配体晶体结构研究了该方法的稳健性,以及63种具有配体的结构多样的晶体结构,适合Twilight数据库中的电子密度。结果表明,BPMD可以成功地将结合姿势不受电子密度支持的化合物与具有明确电子密度的化合物区分开。
更新日期:2020-01-07
中文翻译:
使用结合姿势代谢动力学研究蛋白质晶体结构中的配体稳定性。
正确的蛋白质-配体结合姿势的鉴定在基于结构的药物设计中很重要,对于评估蛋白质-配体结合亲和力至关重要。蛋白质-配体坐标通常从晶体学实验中获得,该晶体学实验提供了构象整体的静态模型。结合姿势元动力学(BPMD)是一种增强的采样方法,可以有效评估溶液中的配体稳定性。在元动力学模拟的偏压下不稳定的配体姿势预计将很少出现在能量图中,因此对结合亲和力的贡献很小。在此,使用已知配体模型错误的配体晶体结构研究了该方法的稳健性,以及63种具有配体的结构多样的晶体结构,适合Twilight数据库中的电子密度。结果表明,BPMD可以成功地将结合姿势不受电子密度支持的化合物与具有明确电子密度的化合物区分开。










































 京公网安备 11010802027423号
京公网安备 11010802027423号