当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
“Disrupt and induce” intermolecular interactions to rationally design organic semiconductor crystals: from herringbone to rubrene-like pitched π-stacking
Chemical Science ( IF 7.6 ) Pub Date : 2020/01/07 , DOI: 10.1039/c9sc05902d Chengyuan Wang 1, 2, 3, 4 , Daisuke Hashizume 2, 3, 4, 5 , Masahiro Nakano 1, 2, 3, 4 , Takuya Ogaki 1, 2, 3, 4 , Hiroyuki Takenaka 4, 6, 7, 8, 9 , Kohsuke Kawabata 4, 6, 7, 8, 9 , Kazuo Takimiya 1, 2, 3, 4, 6
Chemical Science ( IF 7.6 ) Pub Date : 2020/01/07 , DOI: 10.1039/c9sc05902d Chengyuan Wang 1, 2, 3, 4 , Daisuke Hashizume 2, 3, 4, 5 , Masahiro Nakano 1, 2, 3, 4 , Takuya Ogaki 1, 2, 3, 4 , Hiroyuki Takenaka 4, 6, 7, 8, 9 , Kohsuke Kawabata 4, 6, 7, 8, 9 , Kazuo Takimiya 1, 2, 3, 4, 6
Affiliation
|
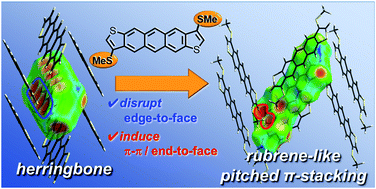
The packing structures of organic semiconductors in the solid state play critical roles in determining the performances of their optoelectronic devices, such as organic field-effect transistors (OFETs). It is a formidable challenge to rationally design molecular packing in the solid state owing to the difficulty of controlling intermolecular interactions. Here we report a unique materials design strategy based on the β-methylthionation of acenedithiophenes to generally and selectively control the packing structures of materials to create organic semiconductors rivalling rubrene, a benchmark high-mobility material with a characteristic pitched π-stacking structure in the solid state. Furthermore, the effect of the β-methylthionation on the packing structure was analyzed by Hirshfeld surface analysis together with theoretical calculations based on symmetry-adapted perturbation theory (SAPT). The results clearly demonstrated that the β-methylthionation of acenedithiophenes can universally alter the intermolecular interactions by disrupting the favorable edge-to-face manner in the parent acenedithiophenes and simultaneously inducing face-to-face and end-to-face interactions in the β-methylthionated acenedithiophenes. This “disrupt and induce” strategy to manipulate intermolecular interactions can open a door to rational packing design based on the molecular structure.
中文翻译:

“破坏并诱导”分子间的相互作用,以合理设计有机半导体晶体:从人字形到类似红荧烯的倾斜π堆积
固态有机半导体的堆积结构在确定其光电器件(例如有机场效应晶体管(OFET))的性能方面起着至关重要的作用。由于难以控制分子间的相互作用,合理设计固态的分子堆积是一个巨大的挑战。在这里,我们报告了一种独特的材料设计策略,该策略基于a庚二噻吩的β-甲基硫磺化来一般性地和选择性地控制材料的堆积结构,以创造出可与红rival烯竞争的有机半导体,而红rub烯是一种在固体中具有特征性的π堆积结构的基准高迁移率材料州。此外,通过Hirshfeld表面分析以及基于对称自适应扰动理论(SAPT)的理论计算,分析了β-甲基硫磺化对堆积结构的影响。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。
更新日期:2020-02-13
中文翻译:
“破坏并诱导”分子间的相互作用,以合理设计有机半导体晶体:从人字形到类似红荧烯的倾斜π堆积
固态有机半导体的堆积结构在确定其光电器件(例如有机场效应晶体管(OFET))的性能方面起着至关重要的作用。由于难以控制分子间的相互作用,合理设计固态的分子堆积是一个巨大的挑战。在这里,我们报告了一种独特的材料设计策略,该策略基于a庚二噻吩的β-甲基硫磺化来一般性地和选择性地控制材料的堆积结构,以创造出可与红rival烯竞争的有机半导体,而红rub烯是一种在固体中具有特征性的π堆积结构的基准高迁移率材料州。此外,通过Hirshfeld表面分析以及基于对称自适应扰动理论(SAPT)的理论计算,分析了β-甲基硫磺化对堆积结构的影响。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。结果清楚地表明,cen苯并噻吩的β-甲硫基化可以通过破坏亲本cen苯并噻吩的有利的边对面方式并同时诱导β-苯并噻吩的面对面和端面相互作用来普遍改变分子间的相互作用。甲基亚硫酰a二噻吩。这种操纵分子间相互作用的“破坏和诱导”策略可以为基于分子结构的合理包装设计打开一扇门。









































 京公网安备 11010802027423号
京公网安备 11010802027423号