当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Flexible Fitting of Biomolecular Structures to Atomic Force Microscopy Images via Biased Molecular Simulations.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-22 , DOI: 10.1021/acs.jctc.9b00991 Toru Niina 1 , Sotaro Fuchigami 1 , Shoji Takada 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-22 , DOI: 10.1021/acs.jctc.9b00991 Toru Niina 1 , Sotaro Fuchigami 1 , Shoji Takada 1
Affiliation
|
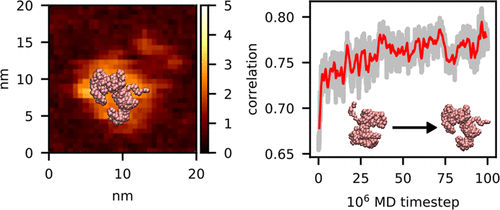
High-speed (HS) atomic force microscopy (AFM) is a prominent imaging technology that observes large-scale structural dynamics of biomolecules near the physiological condition, but the AFM data are limited to the surface shape of specimens. Rigid-body fitting methods were developed to obtain molecular structures that fit to an AFM image, without accounting for conformational changes. Here, we developed a method to fit flexibly a three-dimensional (3D) biomolecular structure into an AFM image. First, we describe a method to produce a pseudo-AFM image from a given 3D structure in a differentiable form. Then, using a correlation function between the experimental AFM image and the computational pseudo-AFM image, we developed a flexible fitting molecular dynamics (MD) simulation method by which we obtain protein structures that well fit to the given AFM image. We first test it with a twin experiment; using an AFM image produced from a protein structure different from its native conformation as a reference, we performed the flexible fitting MD simulations to sample conformations that fit well the reference AFM image, and the method was confirmed to work well. Then, parameter dependence in the protocol was discussed. Finally, we applied the method to a real experimental HS-AFM image for a flagellar protein FlhA, demonstrating its applicability. We also test the rigid-body fitting of a molecular structure to an AFM image. Our method will be a general tool for dynamic structure modeling based on HS-AFM images and is publicly available through the CafeMol software.
中文翻译:

通过偏置分子模拟将生物分子结构灵活地拟合到原子力显微镜图像上。
高速(HS)原子力显微镜(AFM)是一项杰出的成像技术,可在生理条件附近观察生物分子的大规模结构动力学,但AFM数据仅限于标本的表面形状。开发了刚体拟合方法,以获取适合AFM图像的分子结构,而无需考虑构象变化。在这里,我们开发了一种将三维(3D)生物分子结构灵活地适合AFM图像的方法。首先,我们描述了一种以可微分形式从给定3D结构生成伪AFM图像的方法。然后,使用实验AFM图像和计算伪AFM图像之间的相关函数,我们开发了一种灵活的拟合分子动力学(MD)模拟方法,通过该方法,我们可以获得与给定的AFM图像非常吻合的蛋白质结构。我们首先通过双实验对其进行测试;使用由不同于其天然构象的蛋白质结构产生的AFM图像作为参考,我们进行了灵活的拟合MD模拟,以对非常适合参考AFM图像的构象进行采样,并且该方法被证实行之有效。然后,讨论了协议中的参数依赖性。最后,我们将该方法应用于鞭毛蛋白FlhA的真实实验HS-AFM图像,证明了其适用性。我们还测试了分子结构对AFM图像的刚体拟合。
更新日期:2020-01-23
中文翻译:
通过偏置分子模拟将生物分子结构灵活地拟合到原子力显微镜图像上。
高速(HS)原子力显微镜(AFM)是一项杰出的成像技术,可在生理条件附近观察生物分子的大规模结构动力学,但AFM数据仅限于标本的表面形状。开发了刚体拟合方法,以获取适合AFM图像的分子结构,而无需考虑构象变化。在这里,我们开发了一种将三维(3D)生物分子结构灵活地适合AFM图像的方法。首先,我们描述了一种以可微分形式从给定3D结构生成伪AFM图像的方法。然后,使用实验AFM图像和计算伪AFM图像之间的相关函数,我们开发了一种灵活的拟合分子动力学(MD)模拟方法,通过该方法,我们可以获得与给定的AFM图像非常吻合的蛋白质结构。我们首先通过双实验对其进行测试;使用由不同于其天然构象的蛋白质结构产生的AFM图像作为参考,我们进行了灵活的拟合MD模拟,以对非常适合参考AFM图像的构象进行采样,并且该方法被证实行之有效。然后,讨论了协议中的参数依赖性。最后,我们将该方法应用于鞭毛蛋白FlhA的真实实验HS-AFM图像,证明了其适用性。我们还测试了分子结构对AFM图像的刚体拟合。










































 京公网安备 11010802027423号
京公网安备 11010802027423号