当前位置:
X-MOL 学术
›
Environ. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Aluminosilicate Mineralogy and the Sorption of Organic Cations: Interplay between Electrostatic Barriers and Compound Structural Features.
Environmental Science & Technology ( IF 10.8 ) Pub Date : 2020-01-21 , DOI: 10.1021/acs.est.9b06121 William C Jolin 1 , Alissa Richard 2 , Dharni Vasudevan 3 , José A Gascón 2 , Allison A MacKay 4
Environmental Science & Technology ( IF 10.8 ) Pub Date : 2020-01-21 , DOI: 10.1021/acs.est.9b06121 William C Jolin 1 , Alissa Richard 2 , Dharni Vasudevan 3 , José A Gascón 2 , Allison A MacKay 4
Affiliation
|
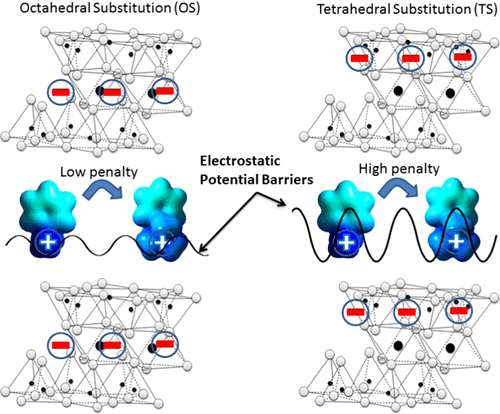
Current predictive models of organic cation sorption assume that sorbates interact with all sites on aluminosilicate minerals in the same manner. To examine whether differences in aluminosilicate structure and the resultant changes in electrostatic potential influence the sorption of organic cations, seven smectites were chosen with different proportions of isomorphic substitutions (origin of clay charge) located in octahedral versus tetrahedral layers and with the presence or absence of aluminosilicate interlayers. Sorption coefficients for 14 benzylamine derivatives with systematic differences in compound structures were collected to understand the possible influence of aluminosilicate mineralogy. Benzylamine compounds with methyl group substitution on the charged amine or with electron-donating or -withdrawing ring substituents displayed decreases in cation exchange-normalized sorption coefficients (KCEC), by up to one order of magnitude, between hectorite (100% isomorphic substitution in the octahedral layer) and nontronite (100% isomorphic substitution in the tetrahedral layer). To understand this difference across aluminosilicates, stochastic molecular models of the various aluminosilicate minerals with interlayers were performed. These models showed that negative charge density associated with tetrahedral sites results in high positive electrostatic energy barriers within the interlayer, creating a penalty for compounds with positive charge spread over a larger compound surface area as occurs from primary to quaternary amines. Conversely, clays with charge originating from octahedral sites produce low electrostatic potential barriers within the interlayer, decreasing the penalty for quaternary amine sorption. Trends for nine cationic pharmaceutical compounds, which varied in size, group alkylation, and/or polar substituents, demonstrated similar decreases in KCEC values to aluminosilicate minerals with high electrostatic energy barriers. Overall, aluminosilicate mineralogy was found to exert a large influence (0.5-1 order of magnitude in sorption coefficients) on organic cation sorption. The application of atomistic electrostatic potential mapping of both sorbent and sorbate structures provided insights to explain trends in sorption coefficients that could not be described by the basic electrostatic potential theory or by assuming that sorbate structure moieties yielded additive sorption contributions.
中文翻译:

铝硅酸盐矿物学与有机阳离子的吸附:静电屏障与复合结构特征之间的相互作用。
当前有机阳离子吸附的预测模型假定吸附物以相同的方式与硅铝酸盐矿物上的所有位点相互作用。为了检查铝硅酸盐结构的差异以及由此产生的静电势的变化是否会影响有机阳离子的吸附,选择了七种蒙脱石,它们分别位于八面体层与四面体层中,同构取代度(粘土电荷的来源)的比例不同,并且存在或不存在铝硅酸盐中间层。收集了在化合物结构上具有系统差异的14种苄胺衍生物的吸附系数,以了解铝硅酸盐矿物学的可能影响。在锂蒙脱石之间(在锂蒙脱石中100%的同构取代),在带电荷的胺上具有甲基取代基或具有给电子或吸电子环取代基的苄胺化合物显示的阳离子交换归一化吸附系数(KCEC)降低多达一个数量级。八面体层)和绿脱石(四面体层中100%的同晶取代)。为了理解铝硅酸盐之间的这种差异,对具有中间层的各种铝硅酸盐矿物进行了随机分子模型研究。这些模型表明,与四面体位点相关的负电荷密度会在中间层内产生较高的正静电能垒,从而对正电荷分布在较大化合物表面积上的化合物(如伯胺和季胺)产生不利影响。相反,带有来自八面体位点的电荷的粘土在中间层内产生较低的静电势垒,从而降低了季胺吸附的代价。九种阳离子药物化合物的趋势,其大小,基团烷基化和/或极性取代基各不相同,显示出KCEC值的降低与具有高静电能垒的硅铝酸盐矿物相似。总体而言,发现铝硅酸盐矿物学对有机阳离子的吸附有很大的影响(吸附系数为0.5-1数量级)。
更新日期:2020-01-22
中文翻译:
铝硅酸盐矿物学与有机阳离子的吸附:静电屏障与复合结构特征之间的相互作用。
当前有机阳离子吸附的预测模型假定吸附物以相同的方式与硅铝酸盐矿物上的所有位点相互作用。为了检查铝硅酸盐结构的差异以及由此产生的静电势的变化是否会影响有机阳离子的吸附,选择了七种蒙脱石,它们分别位于八面体层与四面体层中,同构取代度(粘土电荷的来源)的比例不同,并且存在或不存在铝硅酸盐中间层。收集了在化合物结构上具有系统差异的14种苄胺衍生物的吸附系数,以了解铝硅酸盐矿物学的可能影响。在锂蒙脱石之间(在锂蒙脱石中100%的同构取代),在带电荷的胺上具有甲基取代基或具有给电子或吸电子环取代基的苄胺化合物显示的阳离子交换归一化吸附系数(KCEC)降低多达一个数量级。八面体层)和绿脱石(四面体层中100%的同晶取代)。为了理解铝硅酸盐之间的这种差异,对具有中间层的各种铝硅酸盐矿物进行了随机分子模型研究。这些模型表明,与四面体位点相关的负电荷密度会在中间层内产生较高的正静电能垒,从而对正电荷分布在较大化合物表面积上的化合物(如伯胺和季胺)产生不利影响。相反,带有来自八面体位点的电荷的粘土在中间层内产生较低的静电势垒,从而降低了季胺吸附的代价。九种阳离子药物化合物的趋势,其大小,基团烷基化和/或极性取代基各不相同,显示出KCEC值的降低与具有高静电能垒的硅铝酸盐矿物相似。总体而言,发现铝硅酸盐矿物学对有机阳离子的吸附有很大的影响(吸附系数为0.5-1数量级)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号