Nature Methods ( IF 36.1 ) Pub Date : 2020-01-06 , DOI: 10.1038/s41592-019-0687-1 Joseph M Cunningham 1 , Grigoriy Koytiger 1, 2 , Peter K Sorger 1 , Mohammed AlQuraishi 1
|
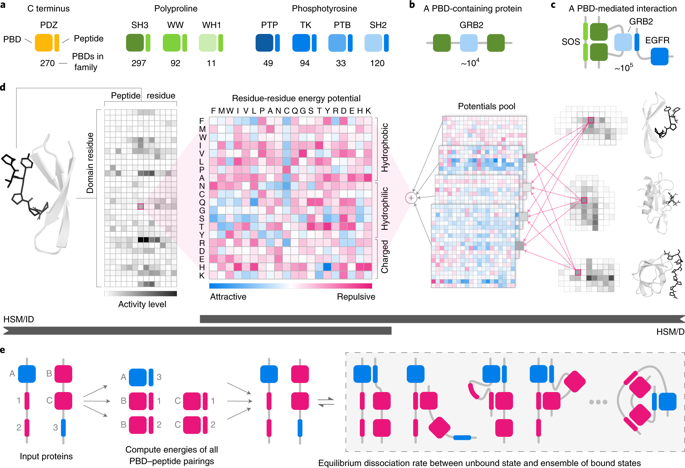
In mammalian cells, much of signal transduction is mediated by weak protein–protein interactions between globular peptide-binding domains (PBDs) and unstructured peptidic motifs in partner proteins. The number and diversity of these PBDs (over 1,800 are known), their low binding affinities and the sensitivity of binding properties to minor sequence variation represent a substantial challenge to experimental and computational analysis of PBD specificity and the networks PBDs create. Here, we introduce a bespoke machine-learning approach, hierarchical statistical mechanical modeling (HSM), capable of accurately predicting the affinities of PBD–peptide interactions across multiple protein families. By synthesizing biophysical priors within a modern machine-learning framework, HSM outperforms existing computational methods and high-throughput experimental assays. HSM models are interpretable in familiar biophysical terms at three spatial scales: the energetics of protein–peptide binding, the multidentate organization of protein–protein interactions and the global architecture of signaling networks.
中文翻译:
使用机器学习对蛋白质-肽相互作用和信号网络进行生物物理预测
在哺乳动物细胞中,大部分信号转导是由球状肽结合域 (PBD) 和伴侣蛋白中的非结构化肽基序之间的弱蛋白-蛋白相互作用介导的。这些 PBD 的数量和多样性(已知超过 1,800 个)、它们的低结合亲和力和结合特性对微小序列变异的敏感性代表了对 PBD 特异性和 PBD 创建的网络的实验和计算分析的重大挑战。在这里,我们介绍了一种定制的机器学习方法,分层统计力学建模 (HSM),能够准确预测 PBD-肽相互作用在多个蛋白质家族中的亲和力。通过在现代机器学习框架内综合生物物理先验,HSM 优于现有的计算方法和高通量实验分析。HSM 模型可以用熟悉的生物物理术语在三个空间尺度上进行解释:蛋白质-肽结合的能量学、蛋白质-蛋白质相互作用的多齿组织和信号网络的全局结构。










































 京公网安备 11010802027423号
京公网安备 11010802027423号