Current Computer-Aided Drug Design ( IF 1.5 ) Pub Date : 2020-09-30 , DOI: 10.2174/1573409915666191025114009 Kauê Santana da Costa 1 , João M Galúcio 1 , Deivid Almeida de Jesus 1 , Guelber Cardoso Gomes 2 , Anderson Henrique Lima E Lima 2 , Paulo S Taube 1 , Alberto M Dos Santos 1 , Jerônimo Lameira 3
|
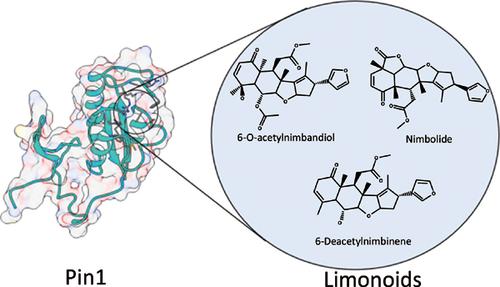
Background: Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) is an enzyme that isomerizes phosphorylated serine or threonine motifs adjacent to proline residues. Pin1 has important roles in several cellular signaling pathways, consequently impacting the development of multiple types of cancers.
Methods: Based on the previously reported inhibitory activity of pentacyclic triterpenoids isolated from the gum resin of Boswellia genus against Pin1, we designed a computational experiment using molecular docking, pharmacophore filtering, and structural clustering allied to molecular dynamics (MD) simulations and binding free energy calculations to explore the inhibitory activity of new triterpenoids against Pin1 structure.
Results: Here, we report different computational evidence that triterpenoids from neem (Azadirachta indica A. Juss), such as 6-deacetylnimbinene, 6-Oacetylnimbandiol, and nimbolide, replicate the binding mode of the Pin1 substrate peptide, interacting with high affinity with the binding site and thus destabilizing the Pin1 structure.
Conclusions: Our results are supported by experimental data, and provide interesting structural insights into their molecular mechanism of action, indicating that their structural scaffolds could be used as a start point to develop new inhibitors against Pin1.
中文翻译:
Targeting Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1: A Structure-based Virtual Screening Approach to Find New Inhibitors。
背景:肽基-脯氨酰顺反异构酶 NIMA 相互作用 1 (Pin1) 是一种酶,可将脯氨酸残基附近的磷酸化丝氨酸或苏氨酸基序异构化。Pin1 在多种细胞信号通路中具有重要作用,因此影响多种类型癌症的发展。
方法:基于先前报道的从乳香属树胶脂中分离的五环三萜类化合物对 Pin1 的抑制活性,我们设计了一个计算实验,使用分子对接、药效团过滤和与分子动力学 (MD) 模拟和结合自由能相关的结构聚类计算以探索新三萜类化合物对 Pin1 结构的抑制活性。
结果:在这里,我们报告了不同的计算证据,表明来自印楝 (Azadirachta indica A. Juss) 的三萜类化合物,如 6-脱乙酰基茚烯、6-邻乙酰茚二醇和茛内酯,复制 Pin1 底物肽的结合模式,以高亲和力与结合位点,从而破坏 Pin1 结构的稳定性。
结论:我们的结果得到了实验数据的支持,并为其分子作用机制提供了有趣的结构见解,表明它们的结构支架可用作开发针对 Pin1 的新抑制剂的起点。










































 京公网安备 11010802027423号
京公网安备 11010802027423号