Nature Methods ( IF 36.1 ) Pub Date : 2018-10-30 , DOI: 10.1038/s41592-018-0173-1 Thomas C Terwilliger 1, 2 , Paul D Adams 3, 4 , Pavel V Afonine 3, 5 , Oleg V Sobolev 3
|
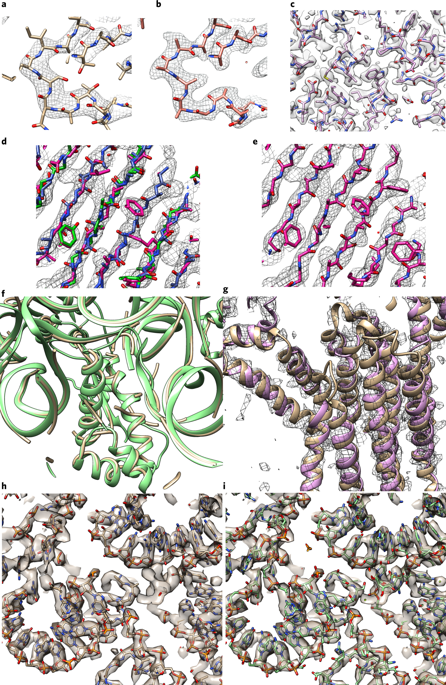
We report a fully automated procedure for the optimization and interpretation of reconstructions from cryo-electron microscopy (cryo-EM) data, available in Phenix as phenix.map_to_model. We applied our approach to 476 datasets with resolution of 4.5 Å or better, including reconstructions of 47 ribosomes and 32 other protein–RNA complexes. The median fraction of residues in the deposited structures reproduced automatically was 71% for reconstructions determined at resolutions of 3 Å or better and 47% for those at resolutions worse than 3 Å.
中文翻译:
一种从高分辨率冷冻电子显微镜图生成初始模型的全自动方法
我们报告了一个全自动程序,用于优化和解释冷冻电子显微镜 (cryo-EM) 数据的重建,可在 Phenix 中作为 phenix.map_to_model 获得。我们将我们的方法应用于 476 个分辨率为 4.5 Å 或更高的数据集,包括 47 个核糖体和 32 个其他蛋白质-RNA 复合物的重建。对于以 3 Å 或更好的分辨率确定的重建,自动再现的沉积结构中残基的中位分数为 71%,对于分辨率低于 3 Å 的重建,残基的中位分数为 47%。










































 京公网安备 11010802027423号
京公网安备 11010802027423号