当前位置:
X-MOL 学术
›
React. Chem. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Revealing quantum mechanical effects in enzyme catalysis with large-scale electronic structure simulation.
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2018-11-29 , DOI: 10.1039/c8re00213d Zhongyue Yang 1 , Rimsha Mehmood 1, 2 , Mengyi Wang 1, 3 , Helena W Qi 1, 2 , Adam H Steeves 1 , Heather J Kulik 1
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2018-11-29 , DOI: 10.1039/c8re00213d Zhongyue Yang 1 , Rimsha Mehmood 1, 2 , Mengyi Wang 1, 3 , Helena W Qi 1, 2 , Adam H Steeves 1 , Heather J Kulik 1
Affiliation
|
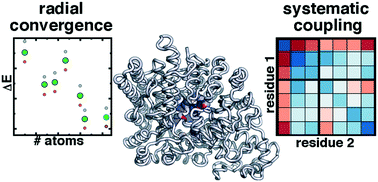
Enzymes have evolved to facilitate challenging reactions at ambient conditions with specificity seldom matched by other catalysts. Computational modeling provides valuable insight into catalytic mechanism, and the large size of enzymes mandates multi-scale, quantum mechanical-molecular mechanical (QM/MM) simulations. Although QM/MM plays an essential role in balancing simulation cost to enable sampling with full QM treatment needed to understand electronic structure in enzyme active sites, the relative importance of these two strategies for understanding enzyme mechanism is not well known. We explore challenges in QM/MM for studying the reactivity and stability of three diverse enzymes: i) Mg2+-dependent catechol O-methyltransferase (COMT), ii) radical enzyme choline trimethylamine lyase (CutC), and iii) DNA methyltransferase (DNMT1), which has structural Zn2+ binding sites. In COMT, strong non-covalent interactions lead to long range coupling of electronic structure properties across the active site, but the more isolated nature of the metallocofactor in DNMT1 leads to faster convergence of some properties. We quantify these effects in COMT by computing covariance matrices of by-residue electronic structure properties during dynamics and along the reaction coordinate. In CutC, we observe spontaneous bond cleavage following initiation events, highlighting the importance of sampling and dynamics. We use electronic structure analysis to quantify the relative importance of CHO and OHO non-covalent interactions in imparting reactivity. These three diverse cases enable us to provide some general recommendations regarding QM/MM simulation of enzymes.
中文翻译:

通过大规模电子结构模拟揭示酶催化中的量子力学效应。
酶已经发展到能够在环境条件下促进具有挑战性的反应,其特异性很少与其他催化剂相匹配。计算模型为催化机制提供了宝贵的见解,而大尺寸的酶需要进行多尺度的量子力学-分子力学 (QM/MM) 模拟。尽管 QM/MM 在平衡模拟成本方面发挥着重要作用,以实现通过全面 QM 处理进行采样以了解酶活性位点的电子结构,但这两种策略对于了解酶机制的相对重要性尚不清楚。我们探索 QM/MM 中研究三种不同酶的反应性和稳定性的挑战:i) Mg2+ 依赖性儿茶酚 O-甲基转移酶 (COMT)、ii) 自由基酶胆碱三甲胺裂解酶 (CutC) 和 iii) DNA 甲基转移酶 (DNMT1) ,其具有结构 Zn2+ 结合位点。在 COMT 中,强非共价相互作用导致整个活性位点电子结构特性的长程耦合,但 DNMT1 中金属辅因子更孤立的性质导致某些特性更快地收敛。我们通过计算动力学期间和沿反应坐标的残基电子结构特性的协方差矩阵来量化 COMT 中的这些影响。在 CutC 中,我们观察到引发事件后自发的键断裂,强调了采样和动力学的重要性。我们使用电子结构分析来量化 CHO 和 OHO 非共价相互作用在赋予反应性方面的相对重要性。这三个不同的案例使我们能够提供一些有关酶的 QM/MM 模拟的一般性建议。
更新日期:2018-11-29
中文翻译:
通过大规模电子结构模拟揭示酶催化中的量子力学效应。
酶已经发展到能够在环境条件下促进具有挑战性的反应,其特异性很少与其他催化剂相匹配。计算模型为催化机制提供了宝贵的见解,而大尺寸的酶需要进行多尺度的量子力学-分子力学 (QM/MM) 模拟。尽管 QM/MM 在平衡模拟成本方面发挥着重要作用,以实现通过全面 QM 处理进行采样以了解酶活性位点的电子结构,但这两种策略对于了解酶机制的相对重要性尚不清楚。我们探索 QM/MM 中研究三种不同酶的反应性和稳定性的挑战:i) Mg2+ 依赖性儿茶酚 O-甲基转移酶 (COMT)、ii) 自由基酶胆碱三甲胺裂解酶 (CutC) 和 iii) DNA 甲基转移酶 (DNMT1) ,其具有结构 Zn2+ 结合位点。在 COMT 中,强非共价相互作用导致整个活性位点电子结构特性的长程耦合,但 DNMT1 中金属辅因子更孤立的性质导致某些特性更快地收敛。我们通过计算动力学期间和沿反应坐标的残基电子结构特性的协方差矩阵来量化 COMT 中的这些影响。在 CutC 中,我们观察到引发事件后自发的键断裂,强调了采样和动力学的重要性。我们使用电子结构分析来量化 CHO 和 OHO 非共价相互作用在赋予反应性方面的相对重要性。这三个不同的案例使我们能够提供一些有关酶的 QM/MM 模拟的一般性建议。










































 京公网安备 11010802027423号
京公网安备 11010802027423号