当前位置:
X-MOL 学术
›
React. Chem. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Decomposition kinetics for HONO and HNO2
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2018-11-29 00:00:00 , DOI: 10.1039/c8re00201k Xi Chen 1, 2, 3, 4 , Mark E. Fuller 2, 3, 4, 5 , C. Franklin Goldsmith 2, 3, 4, 5
Reaction Chemistry & Engineering ( IF 3.4 ) Pub Date : 2018-11-29 00:00:00 , DOI: 10.1039/c8re00201k Xi Chen 1, 2, 3, 4 , Mark E. Fuller 2, 3, 4, 5 , C. Franklin Goldsmith 2, 3, 4, 5
Affiliation
|
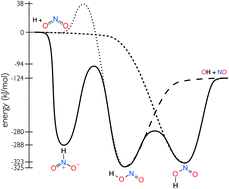
This work presents a detailed investigation into the isomerization and decomposition of HONO and HNO2. State-of-the-art electronic structure theory is used to compute the HNO2 potential energy surface. Temperature and pressure dependent rate coefficients are computed using microcanonical rate theory and the master equation. The electronic structure theory properties are optimized against the relevant experimental data. A novel strategy was developed to incorporate uncertainty in the minimum energy pathway into the optimized mechanism. The new mechanism is in excellent agreement with all available experimental data for H + NO2 → OH + NO and OH + NO → HONO. The calculations identify OH + NO as the dominant products for HNO2, which were neglected from all previous mechanisms in the literature.
中文翻译:

HONO和HNO 2的分解动力学
这项工作提出了对HONO和HNO 2的异构化和分解的详细研究。最新的电子结构理论用于计算HNO 2势能面。使用微规范速率理论和主方程计算温度和压力相关的速率系数。电子结构理论特性针对相关的实验数据进行了优化。开发了一种新策略,将最小能量路径中的不确定性纳入优化机制。该新机制与H + NO 2 →OH + NO和OH + NO→HONO的所有可用实验数据高度吻合。计算确定OH + NO是HNO 2的主要产物,这在文献中所有以前的机制中都被忽略了。
更新日期:2018-11-29
中文翻译:
HONO和HNO 2的分解动力学
这项工作提出了对HONO和HNO 2的异构化和分解的详细研究。最新的电子结构理论用于计算HNO 2势能面。使用微规范速率理论和主方程计算温度和压力相关的速率系数。电子结构理论特性针对相关的实验数据进行了优化。开发了一种新策略,将最小能量路径中的不确定性纳入优化机制。该新机制与H + NO 2 →OH + NO和OH + NO→HONO的所有可用实验数据高度吻合。计算确定OH + NO是HNO 2的主要产物,这在文献中所有以前的机制中都被忽略了。










































 京公网安备 11010802027423号
京公网安备 11010802027423号