当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Calculation of Vibrational Relaxation Times Using a Kinetic Theory Approach
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-11-28 00:00:00 , DOI: 10.1021/acs.jpca.8b09897 G. P. Oblapenko 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-11-28 00:00:00 , DOI: 10.1021/acs.jpca.8b09897 G. P. Oblapenko 1
Affiliation
|
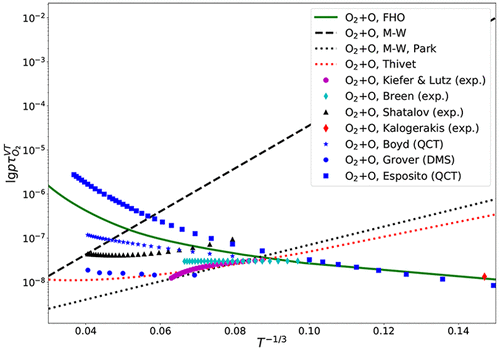
In the present work, a method for computation of vibrational relaxation times based on a kinetic theory definition is utilized to calculate vibrational relaxation times of molecules present in air (N2, O2, and NO) in collisions with air species particles. An overview of available experiment VT relaxation time measurements, as well as quasi-classical trajectory (QCT) calculation results, and various empirical models, is given. Different inelastic cross-section models are used for the computation of the relaxation times, and their parameters are adjusted to fit the available experimental data and QCT results. It is shown that the proposed method of calculation can give a quantitative and qualitative agreement with the available data in a wide range of temperatures; the obtained interaction parameters may be used not only for vibrational relaxation time calculation within a multitemperature framework but also for development of state-specific models for use in CFD and DSMC codes.
中文翻译:

用动力学理论方法计算振动弛豫时间
在本工作中,利用基于动力学理论定义的振动弛豫时间的计算方法来计算空气中存在的分子(N 2,O 2和NO)与空气中的微粒碰撞。给出了可用的实验VT弛豫时间测量以及准经典轨迹(QCT)计算结果和各种经验模型的概述。使用不同的非弹性横截面模型来计算弛豫时间,并调整其参数以适合可用的实验数据和QCT结果。结果表明,所提出的计算方法可以在很宽的温度范围内与现有数据给出定量和定性的一致性。获得的相互作用参数不仅可以用于多温度框架内的振动弛豫时间计算,还可以用于开发用于CFD和DSMC代码的特定于状态的模型。
更新日期:2018-11-28
中文翻译:
用动力学理论方法计算振动弛豫时间
在本工作中,利用基于动力学理论定义的振动弛豫时间的计算方法来计算空气中存在的分子(N 2,O 2和NO)与空气中的微粒碰撞。给出了可用的实验VT弛豫时间测量以及准经典轨迹(QCT)计算结果和各种经验模型的概述。使用不同的非弹性横截面模型来计算弛豫时间,并调整其参数以适合可用的实验数据和QCT结果。结果表明,所提出的计算方法可以在很宽的温度范围内与现有数据给出定量和定性的一致性。获得的相互作用参数不仅可以用于多温度框架内的振动弛豫时间计算,还可以用于开发用于CFD和DSMC代码的特定于状态的模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号