当前位置:
X-MOL 学术
›
ChemMedChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Targeted Synthesis of Complex Spiro[3H‐indole‐3,2′‐pyrrolidin]‐2(1H)‐ones by Intramolecular Cyclization of Azomethine Ylides: Highly Potent MDM2–p53 Inhibitors
ChemMedChem ( IF 3.6 ) Pub Date : 2018-12-11 , DOI: 10.1002/cmdc.201800617 Andreas Gollner 1 , Harald Weinstabl 1 , Julian E Fuchs 1 , Dorothea Rudolph 1 , Geraldine Garavel 1 , Karin S Hofbauer 1 , Jale Karolyi-Oezguer 1 , Gerhard Gmaschitz 1 , Wolfgang Hela 1 , Nina Kerres 1 , Elisabeth Grondal 1 , Patrick Werni 1 , Juergen Ramharter 1 , Joachim Broeker 1 , Darryl B McConnell 1
ChemMedChem ( IF 3.6 ) Pub Date : 2018-12-11 , DOI: 10.1002/cmdc.201800617 Andreas Gollner 1 , Harald Weinstabl 1 , Julian E Fuchs 1 , Dorothea Rudolph 1 , Geraldine Garavel 1 , Karin S Hofbauer 1 , Jale Karolyi-Oezguer 1 , Gerhard Gmaschitz 1 , Wolfgang Hela 1 , Nina Kerres 1 , Elisabeth Grondal 1 , Patrick Werni 1 , Juergen Ramharter 1 , Joachim Broeker 1 , Darryl B McConnell 1
Affiliation
|
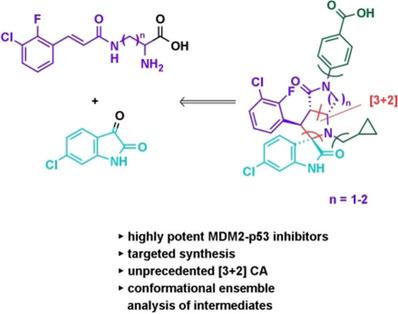
Mouse double minute 2 (MDM2) is a main and direct inhibitor of the crucial tumor suppressor p53. Reports from initial clinical trials showed that blocking this interaction with a small‐molecule inhibitor can have great value in the treatment of cancer for patients with p53 wild‐type tumors; however, it also revealed dose‐limiting hematological toxicities and drug‐induced resistance as main issues. To overcome the former, an inhibitor with superior potency and pharmacokinetic properties to ultimately achieve full efficacy with less‐frequent dosing schedules is required. Toward this aim, we optimized our recently reported spiro‐oxindole inhibitors by focusing on the crucial interaction with the amino acid side chain of His96MDM2. The designed molecules required the targeted synthesis of structurally complex spiro[indole‐3,2′‐pyrrolo[2,3‐c]pyrrole]‐2,4′‐diones for which we developed an unprecedented intramolecular azomethine ylide cycloaddition and investigated the results by computational methods. One of the new compounds showed superior cellular potency over previously reported BI‐0252. This finding is a significant step toward an inhibitor suitable to potentially mitigate hematological on‐target adverse effects.
中文翻译:

通过甲亚碱叶立德分子内环化靶向合成复杂螺[3H-吲哚-3,2′-吡咯烷]-2(1H)-酮:高效MDM2-p53抑制剂
小鼠双分钟 2 (MDM2) 是关键肿瘤抑制因子 p53 的主要和直接抑制剂。初步临床试验报告表明,用小分子抑制剂阻断这种相互作用对于 p53 野生型肿瘤患者的癌症治疗具有巨大价值;然而,它也揭示了剂量限制性血液学毒性和药物诱导的耐药性是主要问题。为了克服前者,需要一种具有卓越效力和药代动力学特性的抑制剂,以最终以较低频率的给药方案实现全部疗效。为了实现这一目标,我们通过关注与 His96 MDM2氨基酸侧链的关键相互作用来优化最近报道的螺羟吲哚抑制剂。设计的分子需要靶向合成结构复杂的螺[吲哚-3,2'-吡咯并[2,3- c ]吡咯]-2,4'-二酮,为此我们开发了前所未有的分子内偶氮甲碱叶立德环加成反应并研究了结果通过计算方法。其中一种新化合物显示出比之前报道的 BI-0252 更优越的细胞效力。这一发现是朝着适合潜在减轻血液学靶向不良反应的抑制剂迈出的重要一步。
更新日期:2018-12-11
中文翻译:
通过甲亚碱叶立德分子内环化靶向合成复杂螺[3H-吲哚-3,2′-吡咯烷]-2(1H)-酮:高效MDM2-p53抑制剂
小鼠双分钟 2 (MDM2) 是关键肿瘤抑制因子 p53 的主要和直接抑制剂。初步临床试验报告表明,用小分子抑制剂阻断这种相互作用对于 p53 野生型肿瘤患者的癌症治疗具有巨大价值;然而,它也揭示了剂量限制性血液学毒性和药物诱导的耐药性是主要问题。为了克服前者,需要一种具有卓越效力和药代动力学特性的抑制剂,以最终以较低频率的给药方案实现全部疗效。为了实现这一目标,我们通过关注与 His96 MDM2氨基酸侧链的关键相互作用来优化最近报道的螺羟吲哚抑制剂。设计的分子需要靶向合成结构复杂的螺[吲哚-3,2'-吡咯并[2,3- c ]吡咯]-2,4'-二酮,为此我们开发了前所未有的分子内偶氮甲碱叶立德环加成反应并研究了结果通过计算方法。其中一种新化合物显示出比之前报道的 BI-0252 更优越的细胞效力。这一发现是朝着适合潜在减轻血液学靶向不良反应的抑制剂迈出的重要一步。










































 京公网安备 11010802027423号
京公网安备 11010802027423号