当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
QTAIM Assessment of the Intra- and Intermolecular Bonding in a Bis(nitramido–oxadiazolate) Energetic Ionic Salt at 20 K
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-11-20 00:00:00 , DOI: 10.1021/acs.jpca.8b10065 Jeremiah P. Tidey 1 , Vladimir V. Zhurov 1 , Christopher G. Gianopoulos 1 , Tobias S. Hermann 2 , A. Alan Pinkerton 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-11-20 00:00:00 , DOI: 10.1021/acs.jpca.8b10065 Jeremiah P. Tidey 1 , Vladimir V. Zhurov 1 , Christopher G. Gianopoulos 1 , Tobias S. Hermann 2 , A. Alan Pinkerton 1
Affiliation
|
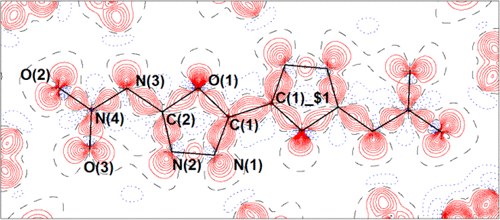
Accurate experimental determination of the electron density distribution for the energetic ionic salt bis(ammonium) 2,2′-dinitramido-5,5′-bis(1-oxa-3,4-diazolate) dihydrate (1) is obtained from multipole modeling of single-crystal X-ray diffraction data collected at 20 K. The intra- and intermolecular bonding is assessed in terms of the quantum theory of atoms in molecules (QTAIM) with a view to better understanding the physicochemical properties in relation to chemical bonding. Topological analysis reveals stronger bonding for the N–NO2 bond relative to energetic nitramines RDX and HMX and the indication of a trend between this and impact sensitivity of nitro-containing energetic materials is noted. The intermolecular bonding of 1 is dominated by classical H-bonds but includes multiple π-bonding interactions and interactions between H-bond donor and acceptor atoms where bond paths are deflected by H atoms. There also exists a weak O···O interaction between end-on nitro groups, as well as an intramolecular ring-forming 1,5-type interaction. An anharmonic description of thermal motion was required to obtain the best fitting model, despite the low temperature of the study. The experimental study was complemented by periodic boundary DFT calculations at the experimental geometry as well as gas phase calculations on the isolated dianion.
中文翻译:

QTAIM评估在20 K下双(硝酰胺基-恶二唑酸酯)高能离子盐中的分子内和分子间键合
通过多极建模获得了高能离子盐双(铵)2,2'-二酰胺基-5,5'-双(1-氧杂-3,4-重氮盐)二水合物(1)的精确电子密度分布的实验确定。在20 K下收集的单晶X射线衍射数据。根据分子中原子的量子理论(QTAIM)评估了分子内和分子间键合,目的是更好地理解与化学键合有关的理化性质。拓扑分析显示,相对于高能硝胺RDX和HMX,N–NO 2键具有更强的键合性,并指出了这种趋势与含硝基高能材料的冲击敏感性之间的趋势。1的分子间键合由经典的H键主导,但包括多个π键相互作用以及H键供体和受体原子之间的相互作用,其中键的路径被H原子偏转。端基硝基之间还存在弱的O··O相互作用,以及分子内形成环的1,5-型相互作用。尽管研究温度较低,但仍需要对热运动进行非谐描述才能获得最佳拟合模型。实验研究得到了实验几何形状的周期性边界DFT计算以及孤立二价阴离子的气相计算的补充。
更新日期:2018-11-20
中文翻译:
QTAIM评估在20 K下双(硝酰胺基-恶二唑酸酯)高能离子盐中的分子内和分子间键合
通过多极建模获得了高能离子盐双(铵)2,2'-二酰胺基-5,5'-双(1-氧杂-3,4-重氮盐)二水合物(1)的精确电子密度分布的实验确定。在20 K下收集的单晶X射线衍射数据。根据分子中原子的量子理论(QTAIM)评估了分子内和分子间键合,目的是更好地理解与化学键合有关的理化性质。拓扑分析显示,相对于高能硝胺RDX和HMX,N–NO 2键具有更强的键合性,并指出了这种趋势与含硝基高能材料的冲击敏感性之间的趋势。1的分子间键合由经典的H键主导,但包括多个π键相互作用以及H键供体和受体原子之间的相互作用,其中键的路径被H原子偏转。端基硝基之间还存在弱的O··O相互作用,以及分子内形成环的1,5-型相互作用。尽管研究温度较低,但仍需要对热运动进行非谐描述才能获得最佳拟合模型。实验研究得到了实验几何形状的周期性边界DFT计算以及孤立二价阴离子的气相计算的补充。










































 京公网安备 11010802027423号
京公网安备 11010802027423号