当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab initio surface hopping excited-state molecular dynamics approach on the basis of spin-orbit coupled states: An application to the A-band photodissociation of CH3 I
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-11-19 , DOI: 10.1002/jcc.25727 Muneaki Kamiya 1 , Tetsuya Taketsugu 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-11-19 , DOI: 10.1002/jcc.25727 Muneaki Kamiya 1 , Tetsuya Taketsugu 2
Affiliation
|
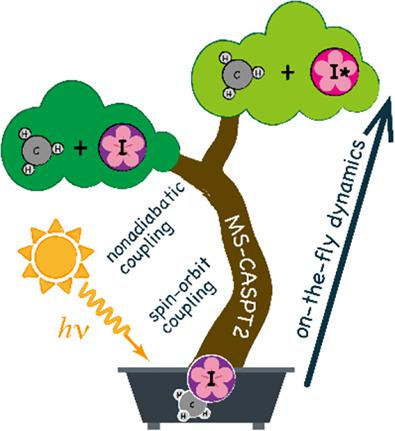
Ab initio molecular dynamics approach has been extended to multi‐state dynamics on the basis of the spin–orbit coupled electronic states that are obtained through diagonalization of the spin–orbit coupling matrix with the multi‐state second‐order multireference perturbation theory energies in diagonal elements and the spin–orbit coupling terms at the state‐averaged complete active space self‐consistent field level in off‐diagonal elements. Nonadiabatic transitions over the spin–orbit coupled states were taken into account explicitly by a surface hopping scheme with utilizing the nonadiabatic coupling terms calculated by numerical differentiation of the spin–orbit coupled wavefunctions and analytical nonadiabatic coupling terms. The present method was applied to the A‐band photodissociation of methyl iodide, CH3I + hv → CH3 + I (2P3/2)/I* (2P1/2), for which a pioneering theoretical work was reported by Amatatsu, Yabushita, and Morokuma. The present results reproduced well the experimental branching ratio and energy distributions in the dissociative products. © 2018 Wiley Periodicals, Inc.
中文翻译:

基于自旋轨道耦合态的从头算表面跳跃激发态分子动力学方法:在 CH3 I 的 A 波段光解中的应用
从头算分子动力学方法已扩展到多态动力学,其基础是自旋轨道耦合电子态,该电子态是通过对角线中的多态二阶多参考微扰理论能量对自旋轨道耦合矩阵进行对角化而获得的。非对角元素中状态平均完整有源空间自洽场能级上的元素和自旋轨道耦合项。表面跳跃方案明确考虑了自旋轨道耦合状态上的非绝热跃迁,利用通过自旋轨道耦合波函数的数值微分和解析非绝热耦合项计算的非绝热耦合项。本方法应用于碘甲烷的 A 波段光解,CH3I + hv → CH3 + I (2P3/2)/I* (2P1/2),Amatatsu、Yabushita 和 Morokuma 报告了一项开创性的理论工作。目前的结果很好地再现了解离产物中的实验支化率和能量分布。© 2018 Wiley Periodicals, Inc.
更新日期:2018-11-19
中文翻译:
基于自旋轨道耦合态的从头算表面跳跃激发态分子动力学方法:在 CH3 I 的 A 波段光解中的应用
从头算分子动力学方法已扩展到多态动力学,其基础是自旋轨道耦合电子态,该电子态是通过对角线中的多态二阶多参考微扰理论能量对自旋轨道耦合矩阵进行对角化而获得的。非对角元素中状态平均完整有源空间自洽场能级上的元素和自旋轨道耦合项。表面跳跃方案明确考虑了自旋轨道耦合状态上的非绝热跃迁,利用通过自旋轨道耦合波函数的数值微分和解析非绝热耦合项计算的非绝热耦合项。本方法应用于碘甲烷的 A 波段光解,CH3I + hv → CH3 + I (2P3/2)/I* (2P1/2),Amatatsu、Yabushita 和 Morokuma 报告了一项开创性的理论工作。目前的结果很好地再现了解离产物中的实验支化率和能量分布。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号