当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Deep Learning for Nonadiabatic Excited-State Dynamics
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-11-07 00:00:00 , DOI: 10.1021/acs.jpclett.8b03026 Wen-Kai Chen 1 , Xiang-Yang Liu 1 , Wei-Hai Fang 1 , Pavlo O. Dral 2 , Ganglong Cui 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-11-07 00:00:00 , DOI: 10.1021/acs.jpclett.8b03026 Wen-Kai Chen 1 , Xiang-Yang Liu 1 , Wei-Hai Fang 1 , Pavlo O. Dral 2 , Ganglong Cui 1
Affiliation
|
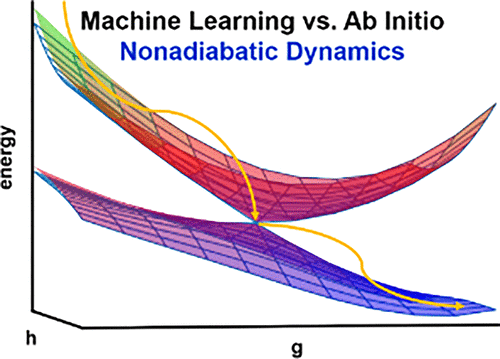
In this work we show that deep learning (DL) can be used for exploring complex and highly nonlinear multistate potential energy surfaces of polyatomic molecules and related nonadiabatic dynamics. Our DL is based on deep neural networks (DNNs), which are used as accurate representations of the CASSCF ground- and excited-state potential energy surfaces (PESs) of CH2NH. After geometries near conical intersection are included in the training set, the DNN models accurately reproduce excited-state topological structures; photoisomerization paths; and, importantly, conical intersections. We have also demonstrated that the results from nonadiabatic dynamics run with the DNN models are very close to those from the dynamics run with the pure ab initio method. The present work should encourage further studies of using machine learning methods to explore excited-state potential energy surfaces and nonadiabatic dynamics of polyatomic molecules.
中文翻译:

非绝热激发态动力学的深度学习
在这项工作中,我们表明深度学习(DL)可用于探索多原子分子的复杂且高度非线性的多态势能面以及相关的非绝热动力学。我们的DL基于深度神经网络(DNN),可作为CH 2的CASSCF基态和激发态势能面(PES)的精确表示。NH。将圆锥形相交点附近的几何形状包括在训练集中之后,DNN模型可以准确地重现激发态拓扑结构;光异构化路径;重要的是圆锥形交叉点。我们还证明,使用DNN模型运行的非绝热动力学结果与使用纯Ab initio方法运行的动力学结果非常接近。当前的工作应鼓励进一步研究使用机器学习方法来探索激发态势能表面和多原子分子的非绝热动力学。
更新日期:2018-11-07
中文翻译:
非绝热激发态动力学的深度学习
在这项工作中,我们表明深度学习(DL)可用于探索多原子分子的复杂且高度非线性的多态势能面以及相关的非绝热动力学。我们的DL基于深度神经网络(DNN),可作为CH 2的CASSCF基态和激发态势能面(PES)的精确表示。NH。将圆锥形相交点附近的几何形状包括在训练集中之后,DNN模型可以准确地重现激发态拓扑结构;光异构化路径;重要的是圆锥形交叉点。我们还证明,使用DNN模型运行的非绝热动力学结果与使用纯Ab initio方法运行的动力学结果非常接近。当前的工作应鼓励进一步研究使用机器学习方法来探索激发态势能表面和多原子分子的非绝热动力学。






































 京公网安备 11010802027423号
京公网安备 11010802027423号