当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Performance of density functional theory for describing hetero-metallic active-site motifs for methane-to-methanol conversion in metal-exchanged zeolites
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-31 , DOI: 10.1002/jcc.25714 Naveen K. Dandu 1 , Olajumoke Adeyiga 1 , Dipak Panthi 1 , Shaina A. Bird 1 , Samuel O. Odoh 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-10-31 , DOI: 10.1002/jcc.25714 Naveen K. Dandu 1 , Olajumoke Adeyiga 1 , Dipak Panthi 1 , Shaina A. Bird 1 , Samuel O. Odoh 1
Affiliation
|
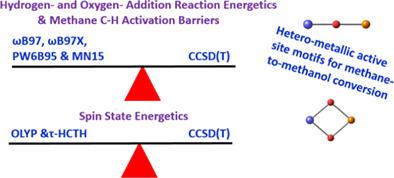
Methane‐to‐methanol conversion (MMC) can be facilitated with high methanol selectivities by copper‐exchanged zeolites. There are however two open questions regarding the use of these zeolites to facilitate the MMC process. The first concerns the possibility of operating the three cycles in the stepwise MMC process by these zeolites in an isothermal fashion. The second concerns the possibility of improving the methanol yields by systematic substitution of some copper centers in these active sites with other earth‐abundant transition metals. Quantum‐mechanical computations can be used to compare methane activation by copper oxide species and analogous mixed‐metal systems. To carry out such screening, it is important that we use theoretical methods that are accurate and computationally affordable for describing the properties of the hetero‐metallic catalytic species. We have examined the performance of 47 exchange‐correlation density functionals for predicting the relative spin‐state energies and chemical reactivities of six hetero‐metallic [M‐O‐Cu]2+ and [M‐O2‐Cu]2+, (where MCo, Fe, and Ni), species by comparison with coupled cluster theory including iterative single, double excitations as well as perturbative treatment of triple excitations, CCSD(T). We also performed multireference calculations on some of these systems. We considered two types of reactions (hydrogen addition and oxygen addition) that are relevant to MMC. We recommend the use of τ‐HCTH and OLYP to determine the spin‐state energy splittings in the hetero‐metallic motifs. ωB97, ωB97X, ωB97X‐D3, and MN15 performed best for predicting the energies of the hydrogen and oxygen addition reactions. In contrast, local, and semilocal functionals do poorly for chemical reactivity. Using [Fe‐O‐Cu]2+ as a test, we see that the nonlocal functionals perform well for the methane CH activation barrier. In contrast, the semilocal functionals perform rather poorly. © 2018 Wiley Periodicals, Inc.
中文翻译:

描述金属交换沸石中甲烷到甲醇转化的异金属活性位点基序的密度泛函理论的性能
铜交换沸石可以通过高甲醇选择性促进甲烷到甲醇的转化(MMC)。然而,关于使用这些沸石促进 MMC 工艺,存在两个悬而未决的问题。第一个涉及通过这些沸石以等温方式在逐步 MMC 过程中操作三个循环的可能性。第二个涉及通过用地球上其他丰富的过渡金属系统取代这些活性位点中的一些铜中心来提高甲醇产率的可能性。量子力学计算可用于比较氧化铜物种和类似混合金属系统对甲烷的活化。为了进行这样的筛选,重要的是我们使用准确且计算上负担得起的理论方法来描述异金属催化物质的性质。我们研究了 47 个交换相关密度泛函的性能,用于预测六种异金属 [M-O-Cu]2+ 和 [M-O2-Cu]2+ 的相对自旋态能量和化学反应性,(其中MCo、Fe 和 Ni),与耦合簇理论相比的物种,包括迭代单、双激发以及三激发的微扰处理 CCSD(T)。我们还对其中一些系统进行了多参考计算。我们考虑了与 MMC 相关的两种类型的反应(加氢和加氧)。我们建议使用 τ-HCTH 和 OLYP 来确定异金属基序中的自旋态能量分裂。ωB97、ωB97X、ωB97X-D3 和 MN15 在预测氢和氧加成反应的能量方面表现最佳。相比之下,局部和半局部泛函在化学反应性方面表现不佳。使用 [Fe-O-Cu]2+ 作为测试,我们看到非局部泛函对于甲烷 CH 激活势垒表现良好。相比之下,半局部泛函的表现相当差。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-31
中文翻译:
描述金属交换沸石中甲烷到甲醇转化的异金属活性位点基序的密度泛函理论的性能
铜交换沸石可以通过高甲醇选择性促进甲烷到甲醇的转化(MMC)。然而,关于使用这些沸石促进 MMC 工艺,存在两个悬而未决的问题。第一个涉及通过这些沸石以等温方式在逐步 MMC 过程中操作三个循环的可能性。第二个涉及通过用地球上其他丰富的过渡金属系统取代这些活性位点中的一些铜中心来提高甲醇产率的可能性。量子力学计算可用于比较氧化铜物种和类似混合金属系统对甲烷的活化。为了进行这样的筛选,重要的是我们使用准确且计算上负担得起的理论方法来描述异金属催化物质的性质。我们研究了 47 个交换相关密度泛函的性能,用于预测六种异金属 [M-O-Cu]2+ 和 [M-O2-Cu]2+ 的相对自旋态能量和化学反应性,(其中MCo、Fe 和 Ni),与耦合簇理论相比的物种,包括迭代单、双激发以及三激发的微扰处理 CCSD(T)。我们还对其中一些系统进行了多参考计算。我们考虑了与 MMC 相关的两种类型的反应(加氢和加氧)。我们建议使用 τ-HCTH 和 OLYP 来确定异金属基序中的自旋态能量分裂。ωB97、ωB97X、ωB97X-D3 和 MN15 在预测氢和氧加成反应的能量方面表现最佳。相比之下,局部和半局部泛函在化学反应性方面表现不佳。使用 [Fe-O-Cu]2+ 作为测试,我们看到非局部泛函对于甲烷 CH 激活势垒表现良好。相比之下,半局部泛函的表现相当差。© 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号