当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fully optimized implementation of the cluster-in-molecule local correlation approach for electron correlation calculations of large systems
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-10-26 , DOI: 10.1002/jcc.25730 Zhigang Ni 1 , Wei Li 1 , Shuhua Li 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-10-26 , DOI: 10.1002/jcc.25730 Zhigang Ni 1 , Wei Li 1 , Shuhua Li 1
Affiliation
|
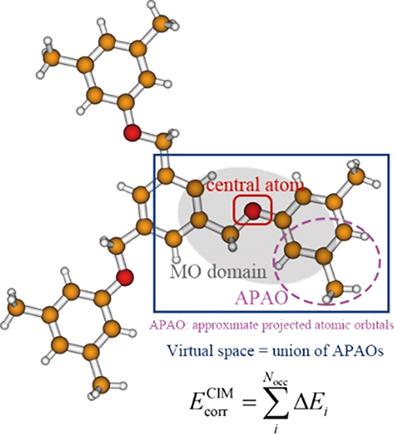
A fully optimized implementation of the cluster‐in‐molecule (CIM) local correlation method for faster and more accurate electron correlation calculations of large systems is reported. The speedup comes from the new procedure of constructing virtual localized molecular orbitals of clusters. In the new procedure, Boughton–Pulay projection method is employed instead of a much more expensive Boys localization procedure. In addition, basis set superposition error correction for binding energy calculations and parallelized electron correlation calculations of clusters are now implemented. Benchmark calculations and illustrative applications at the Møller–Plesset perturbation theory, coupled cluster singles and doubles (CCSD), and CCSD with perturbative triples correction levels show that this newly optimized CIM approach is a reliable theoretical tool for electron correlation calculations of various large chemical systems. © 2018 Wiley Periodicals, Inc.
中文翻译:

用于大型系统电子相关计算的分子簇局部相关方法的完全优化实现
报告了分子簇(CIM)局部相关方法的完全优化实现,用于更快、更准确地计算大型系统的电子相关性。加速来自构建簇的虚拟局部分子轨道的新程序。在新程序中,使用了 Boughton-Pulay 投影方法,而不是更昂贵的 Boys 本地化程序。此外,现在实现了用于结合能计算和并行电子相关计算的基组叠加误差校正。Møller-Plesset 扰动理论的基准计算和说明性应用,耦合簇单打和双打(CCSD),和具有微扰三元组校正水平的 CCSD 表明,这种新优化的 CIM 方法是各种大型化学系统电子相关计算的可靠理论工具。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-26
中文翻译:
用于大型系统电子相关计算的分子簇局部相关方法的完全优化实现
报告了分子簇(CIM)局部相关方法的完全优化实现,用于更快、更准确地计算大型系统的电子相关性。加速来自构建簇的虚拟局部分子轨道的新程序。在新程序中,使用了 Boughton-Pulay 投影方法,而不是更昂贵的 Boys 本地化程序。此外,现在实现了用于结合能计算和并行电子相关计算的基组叠加误差校正。Møller-Plesset 扰动理论的基准计算和说明性应用,耦合簇单打和双打(CCSD),和具有微扰三元组校正水平的 CCSD 表明,这种新优化的 CIM 方法是各种大型化学系统电子相关计算的可靠理论工具。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号