当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluation of Advanced Precursor Determination for Tandem Mass Tag (TMT)-Based Quantitative Proteomics across Instrument Platforms.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-31 , DOI: 10.1021/acs.jproteome.8b00611 Samuel A Myers 1 , Susan Klaeger 1 , Shankha Satpathy 1 , Rosa Viner 2 , Jae Choi 3 , John Rogers 3 , Karl Clauser 1 , Namrata D Udeshi 1 , Steven A Carr 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-31 , DOI: 10.1021/acs.jproteome.8b00611 Samuel A Myers 1 , Susan Klaeger 1 , Shankha Satpathy 1 , Rosa Viner 2 , Jae Choi 3 , John Rogers 3 , Karl Clauser 1 , Namrata D Udeshi 1 , Steven A Carr 1
Affiliation
|
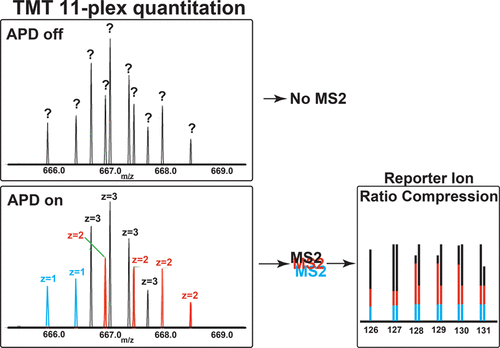
Tandem mass tag (TMT)-based quantitation is a strong modality for quantitative proteomics, as samples can be multiplexed, creating large-scale data sets with high precision and minimal missing values. However, coisolation/cofragmentation of near isobaric, coeluting precursor peptide analytes has been well-documented to show ratio compression, compromising the accuracy of peptide/protein quantitation. Advanced peak determination (APD) is a new peak-picking algorithm that shows improved identification of peak detection in survey scans (MS1) to increase the number of precursors selected for unimolecular dissociation (MS2). To increase the number of these “features” selected for MS2 APD purposefully selects multiple peptide precursors of very similar m/z that often derive from different proteins—a major source of ratio compression in TMT quantification. Here, we evaluate the effects of various data acquisition parameters combined with APD on ratio compression. We find that data acquisition with APD enabled results in more coisolated precursors, more mixed spectra, and in turn, fewer peptide spectral matches, especially at standard on-column loads. We conclude that APD should not be utilized for isobaric tagging, MS2-based experiments.
中文翻译:

评估跨平台的基于串联质量标签(TMT)的定量蛋白质组学的高级前体测定。
基于串联质量标签(TMT)的定量方法是定量蛋白质组学的强大方法,因为可以对样品进行多路复用,从而创建具有高精度和最小缺失值的大规模数据集。然而,近等压共洗脱的前体肽分析物的共分离/共碎化已得到充分证明,显示出比率压缩,从而损害了肽/蛋白质定量的准确性。高级峰测定(APD)是一种新的峰提取算法,该算法显示了改进的调查扫描(MS1)中峰检测的识别能力,从而增加了为单分子解离(MS2)选择的前体数量。为了增加为MS2 APD选择的这些“功能”的数量,有目的地选择m / z非常相似的多种肽前体通常来自不同的蛋白质,这是TMT定量分析中比率压缩的主要来源。在这里,我们评估了与APD结合使用的各种数据采集参数对比率压缩的影响。我们发现,启用APD的数据采集可产生更多的共隔离的前体,更多的混合光谱,进而减少肽段的光谱匹配,尤其是在标准柱上负载下。我们得出结论,APD不应用于基于MS2的等压标记。
更新日期:2018-11-02
中文翻译:
评估跨平台的基于串联质量标签(TMT)的定量蛋白质组学的高级前体测定。
基于串联质量标签(TMT)的定量方法是定量蛋白质组学的强大方法,因为可以对样品进行多路复用,从而创建具有高精度和最小缺失值的大规模数据集。然而,近等压共洗脱的前体肽分析物的共分离/共碎化已得到充分证明,显示出比率压缩,从而损害了肽/蛋白质定量的准确性。高级峰测定(APD)是一种新的峰提取算法,该算法显示了改进的调查扫描(MS1)中峰检测的识别能力,从而增加了为单分子解离(MS2)选择的前体数量。为了增加为MS2 APD选择的这些“功能”的数量,有目的地选择m / z非常相似的多种肽前体通常来自不同的蛋白质,这是TMT定量分析中比率压缩的主要来源。在这里,我们评估了与APD结合使用的各种数据采集参数对比率压缩的影响。我们发现,启用APD的数据采集可产生更多的共隔离的前体,更多的混合光谱,进而减少肽段的光谱匹配,尤其是在标准柱上负载下。我们得出结论,APD不应用于基于MS2的等压标记。










































 京公网安备 11010802027423号
京公网安备 11010802027423号