当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An improved methodology to compute surface site interaction points using high density molecular electrostatic potential surfaces
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25574 Antoni Oliver 1 , Christopher A Hunter 2 , Rafel Prohens 3 , Josep Lluis Rosselló 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-10-23 , DOI: 10.1002/jcc.25574 Antoni Oliver 1 , Christopher A Hunter 2 , Rafel Prohens 3 , Josep Lluis Rosselló 1
Affiliation
|
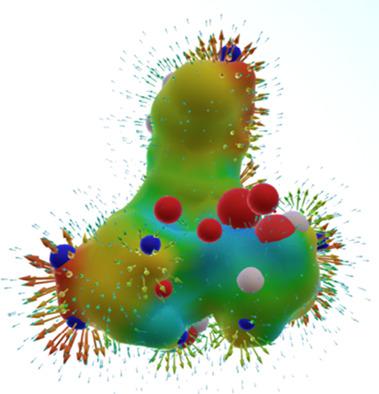
The theoretical calculation of Surface Site Interaction Points (SSIP) has been used successfully in some applications in the solid and liquid phase. In this work we propose a new set of optimizations for the search of SSIP using the Molecular Electrostatic Potential Surfaces (MEPS) calculated with Density Functional Theory and B3LYP/6‐31*G basis set. The measures that have been implemented are based on the search for the best agreement between experimental H‐bond donor and acceptor parameters (α and β) and the MEPS extremes exploring a range of electron density levels. Additionally, a parameterization as a function of atom types has been performed. The results show that the MEPS calculated at 0.01 au electron density level slightly improves the correlation with experimental data in comparison with the calculation over other density values. This fact is related to the bigger contribution of local electrostatics at higher density levels. The refinement has provided significant improvements to the correlation between theoretical and experimental data. Moreover, the proposed calculation over 0.01 au is six times faster on average than the computation at 0.002 au. The proposed methodology has been developed with the purpose to obtain high precision SSIP in a fast way and to improve their applications in virtual cocrystal screening, calculation of free energies in solution and molecular docking. © 2018 Wiley Periodicals, Inc.
中文翻译:

一种使用高密度分子静电势面计算表面位点相互作用点的改进方法
表面位点相互作用点 (SSIP) 的理论计算已成功用于固相和液相的某些应用。在这项工作中,我们使用密度泛函理论和 B3LYP/6-31*G 基组计算的分子静电势面 (MEPS) 提出了一组新的优化,用于搜索 SSIP。已实施的措施基于寻找实验性氢键供体和受体参数(α 和 β)与 MEPS 极端值之间的最佳一致性,探索一系列电子密度水平。此外,还执行了作为原子类型函数的参数化。结果表明,与其他密度值的计算相比,在 0.01 au 电子密度水平计算的 MEPS 略微提高了与实验数据的相关性。这一事实与较高密度水平下局部静电的更大贡献有关。改进显着改善了理论和实验数据之间的相关性。此外,在 0.01 au 上的建议计算平均比在 0.002 au 上的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc. 01 au 平均比 0.002 au 的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc. 01 au 平均比 0.002 au 的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc.
更新日期:2018-10-23
中文翻译:
一种使用高密度分子静电势面计算表面位点相互作用点的改进方法
表面位点相互作用点 (SSIP) 的理论计算已成功用于固相和液相的某些应用。在这项工作中,我们使用密度泛函理论和 B3LYP/6-31*G 基组计算的分子静电势面 (MEPS) 提出了一组新的优化,用于搜索 SSIP。已实施的措施基于寻找实验性氢键供体和受体参数(α 和 β)与 MEPS 极端值之间的最佳一致性,探索一系列电子密度水平。此外,还执行了作为原子类型函数的参数化。结果表明,与其他密度值的计算相比,在 0.01 au 电子密度水平计算的 MEPS 略微提高了与实验数据的相关性。这一事实与较高密度水平下局部静电的更大贡献有关。改进显着改善了理论和实验数据之间的相关性。此外,在 0.01 au 上的建议计算平均比在 0.002 au 上的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc. 01 au 平均比 0.002 au 的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc. 01 au 平均比 0.002 au 的计算快六倍。所提出的方法的开发旨在快速获得高精度 SSIP 并改进其在虚拟共晶筛选、溶液中自由能计算和分子对接中的应用。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号