当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multiple-Enzyme-Digestion Strategy Improves Accuracy and Sensitivity of Label- and Standard-Free Absolute Quantification to a Level That Is Achievable by Analysis with Stable Isotope-Labeled Standard Spiking.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-30 , DOI: 10.1021/acs.jproteome.8b00549 Jacek R Wiśniewski 1 , Christine Wegler 2, 3 , Per Artursson 2, 4
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-30 , DOI: 10.1021/acs.jproteome.8b00549 Jacek R Wiśniewski 1 , Christine Wegler 2, 3 , Per Artursson 2, 4
Affiliation
|
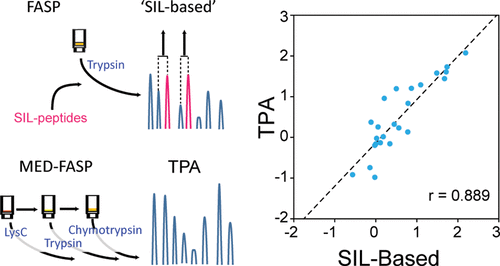
Quantification of individual proteins is an essential task in understanding biological processes. For example, determination of concentrations of proteins transporting and metabolizing xenobiotics is a prerequisite for drug disposition predictions in humans based on in vitro data. So far, this task has frequently been accomplished by targeted proteomics. This type of analyses requires preparation of stable isotope labeled standards for each protein of interest. The selection of appropriate standard peptides is usually tedious and the number of proteins that can be studied in a single experiment by these approaches is limited. In addition, incomplete digestion of proteins often affects the accuracy of the quantification. To circumvent these constrains in proteomic protein quantification, label- and standard-free approaches, such as "total protein approach" (TPA) have been proposed. Here we directly compare an approach using stable isotope labeled (SIL) standards and TPA for quantification of transporters and enzymes in human liver samples within the same LC-MS/MS runs. We show that TPA is a convenient alternative to SIL-based methods. Optimization of the sample preparation beyond commonly used single tryptic digestion, by adding consecutive cleavage steps, improves accuracy and reproducibility of the TPA method to a level, which is achievable by analysis using stable isotope-labeled standard spiking.
中文翻译:

多种酶消化策略将无标签和无标准品的绝对定量的准确性和灵敏度提高到可以通过稳定同位素标记的标准加标分析实现的水平。
单个蛋白质的定量是理解生物学过程的基本任务。例如,基于体外数据,确定运输和代谢异源生物蛋白的浓度是预测人类药物处置的先决条件。到目前为止,该任务通常是通过靶向蛋白质组学来完成的。这种类型的分析需要为每种目标蛋白质制备稳定的同位素标记的标准品。合适的标准肽的选择通常很繁琐,并且可以通过这些方法在单个实验中研究的蛋白质数量有限。另外,蛋白质的不完全消化通常会影响定量的准确性。为了规避蛋白质组蛋白定量,无标签和无标准方法的这些限制,例如“ 我们表明,TPA是基于SIL的方法的方便替代方法。通过添加连续的裂解步骤,除了常用的单个胰蛋白酶消化之外,样品制备的优化还可以将TPA方法的准确性和可重复性提高到一定水平,这可以通过使用稳定的同位素标记的标准加标进行分析来实现。我们表明,TPA是基于SIL的方法的方便替代方法。通过添加连续的裂解步骤,可以优化样品制备方法,使其超出常用的单个胰蛋白酶消化方法,从而将TPA方法的准确性和可重复性提高到一定水平,这可以通过使用稳定的同位素标记的标准加标进行分析来实现。
更新日期:2018-10-31
中文翻译:
多种酶消化策略将无标签和无标准品的绝对定量的准确性和灵敏度提高到可以通过稳定同位素标记的标准加标分析实现的水平。
单个蛋白质的定量是理解生物学过程的基本任务。例如,基于体外数据,确定运输和代谢异源生物蛋白的浓度是预测人类药物处置的先决条件。到目前为止,该任务通常是通过靶向蛋白质组学来完成的。这种类型的分析需要为每种目标蛋白质制备稳定的同位素标记的标准品。合适的标准肽的选择通常很繁琐,并且可以通过这些方法在单个实验中研究的蛋白质数量有限。另外,蛋白质的不完全消化通常会影响定量的准确性。为了规避蛋白质组蛋白定量,无标签和无标准方法的这些限制,例如“ 我们表明,TPA是基于SIL的方法的方便替代方法。通过添加连续的裂解步骤,除了常用的单个胰蛋白酶消化之外,样品制备的优化还可以将TPA方法的准确性和可重复性提高到一定水平,这可以通过使用稳定的同位素标记的标准加标进行分析来实现。我们表明,TPA是基于SIL的方法的方便替代方法。通过添加连续的裂解步骤,可以优化样品制备方法,使其超出常用的单个胰蛋白酶消化方法,从而将TPA方法的准确性和可重复性提高到一定水平,这可以通过使用稳定的同位素标记的标准加标进行分析来实现。










































 京公网安备 11010802027423号
京公网安备 11010802027423号