Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Genome organization and DNA accessibility control antigenic variation in trypanosomes
Nature ( IF 64.8 ) Pub Date : 2018-10-17 , DOI: 10.1038/s41586-018-0619-8 Laura S M Müller 1, 2, 3 , Raúl O Cosentino 1, 2, 3 , Konrad U Förstner 4, 5, 6 , Julien Guizetti 3, 7 , Carolin Wedel 3 , Noam Kaplan 8 , Christian J Janzen 9 , Panagiota Arampatzi 6 , Jörg Vogel 10, 11 , Sascha Steinbiss 12 , Thomas D Otto 12, 13 , Antoine-Emmanuel Saliba 10 , Robert P Sebra 14 , T Nicolai Siegel 1, 2, 3
Nature ( IF 64.8 ) Pub Date : 2018-10-17 , DOI: 10.1038/s41586-018-0619-8 Laura S M Müller 1, 2, 3 , Raúl O Cosentino 1, 2, 3 , Konrad U Förstner 4, 5, 6 , Julien Guizetti 3, 7 , Carolin Wedel 3 , Noam Kaplan 8 , Christian J Janzen 9 , Panagiota Arampatzi 6 , Jörg Vogel 10, 11 , Sascha Steinbiss 12 , Thomas D Otto 12, 13 , Antoine-Emmanuel Saliba 10 , Robert P Sebra 14 , T Nicolai Siegel 1, 2, 3
Affiliation
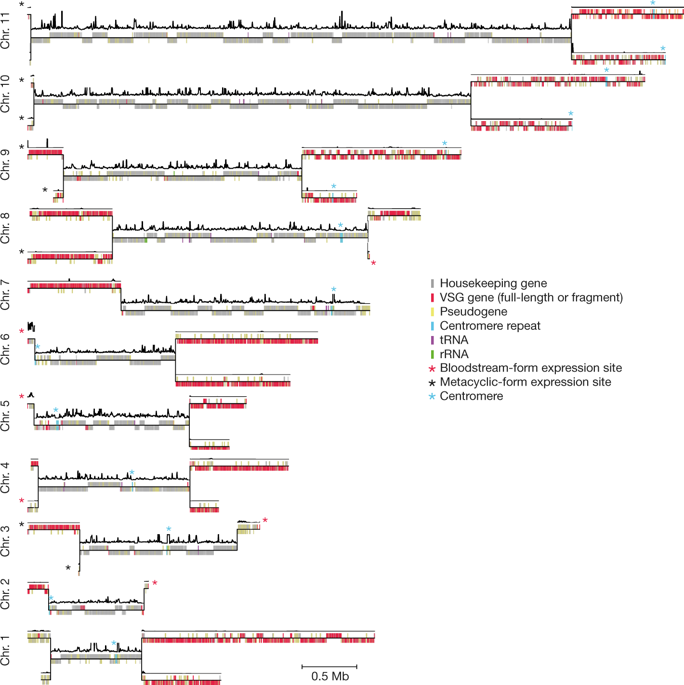
|
Many evolutionarily distant pathogenic organisms have evolved similar survival strategies to evade the immune responses of their hosts. These include antigenic variation, through which an infecting organism prevents clearance by periodically altering the identity of proteins that are visible to the immune system of the host1. Antigenic variation requires large reservoirs of immunologically diverse antigen genes, which are often generated through homologous recombination, as well as mechanisms to ensure the expression of one or very few antigens at any given time. Both homologous recombination and gene expression are affected by three-dimensional genome architecture and local DNA accessibility2,3. Factors that link three-dimensional genome architecture, local chromatin conformation and antigenic variation have, to our knowledge, not yet been identified in any organism. One of the major obstacles to studying the role of genome architecture in antigenic variation has been the highly repetitive nature and heterozygosity of antigen-gene arrays, which has precluded complete genome assembly in many pathogens. Here we report the de novo haplotype-specific assembly and scaffolding of the long antigen-gene arrays of the model protozoan parasite Trypanosoma brucei, using long-read sequencing technology and conserved features of chromosome folding4. Genome-wide chromosome conformation capture (Hi-C) reveals a distinct partitioning of the genome, with antigen-encoding subtelomeric regions that are folded into distinct, highly compact compartments. In addition, we performed a range of analyses—Hi-C, fluorescence in situ hybridization, assays for transposase-accessible chromatin using sequencing and single-cell RNA sequencing—that showed that deletion of the histone variants H3.V and H4.V increases antigen-gene clustering, DNA accessibility across sites of antigen expression and switching of the expressed antigen isoform, via homologous recombination. Our analyses identify histone variants as a molecular link between global genome architecture, local chromatin conformation and antigenic variation.Long-read sequencing allows the assembly of antigen-gene arrays in Trypanosoma brucei and, coupled with deletion experiments, demonstrates that histone variants act as a molecular link between genome architecture, chromatin conformation and antigen variation.
中文翻译:

基因组组织和 DNA 可及性控制锥虫中的抗原变异
许多进化上遥远的病原生物进化出类似的生存策略来逃避宿主的免疫反应。这些包括抗原变异,通过这种变异,感染生物体通过定期改变对宿主免疫系统可见的蛋白质的身份来防止清除。抗原变异需要大量的免疫多样性抗原基因库,这些抗原基因通常是通过同源重组产生的,以及确保在任何给定时间表达一种或很少抗原的机制。同源重组和基因表达都受到三维基因组结构和局部 DNA 可及性的影响 2,3。据我们所知,将三维基因组结构、局部染色质构象和抗原变异联系起来的因素有:尚未在任何生物体中发现。研究基因组结构在抗原变异中的作用的主要障碍之一是抗原基因阵列的高度重复性和杂合性,这阻碍了许多病原体的完整基因组组装。在这里,我们报告了模型原生动物寄生虫布氏锥虫的长抗原基因阵列的从头单倍型特异性组装和支架,使用长读测序技术和染色体折叠的保守特征4。全基因组染色体构象捕获 (Hi-C) 揭示了基因组的不同分区,抗原编码亚端粒区域折叠成不同的、高度紧凑的隔室。此外,我们还进行了一系列分析——Hi-C、荧光原位杂交、使用测序和单细胞 RNA 测序对转座酶可及的染色质进行分析——这表明组蛋白变体 H3.V 和 H4.V 的缺失增加了抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换, 通过同源重组。我们的分析将组蛋白变体鉴定为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并结合缺失实验,证明组蛋白变体作为基因组结构、染色质构象和抗原变异之间的分子联系。V 通过同源重组增加抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换。我们的分析将组蛋白变异识别为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并与缺失实验相结合,证明组蛋白变异可作为基因组结构、染色质构象和抗原变异之间的分子联系。V 通过同源重组增加抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换。我们的分析将组蛋白变体鉴定为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并结合缺失实验,证明组蛋白变体作为基因组结构、染色质构象和抗原变异之间的分子联系。
更新日期:2018-10-17
中文翻译:
基因组组织和 DNA 可及性控制锥虫中的抗原变异
许多进化上遥远的病原生物进化出类似的生存策略来逃避宿主的免疫反应。这些包括抗原变异,通过这种变异,感染生物体通过定期改变对宿主免疫系统可见的蛋白质的身份来防止清除。抗原变异需要大量的免疫多样性抗原基因库,这些抗原基因通常是通过同源重组产生的,以及确保在任何给定时间表达一种或很少抗原的机制。同源重组和基因表达都受到三维基因组结构和局部 DNA 可及性的影响 2,3。据我们所知,将三维基因组结构、局部染色质构象和抗原变异联系起来的因素有:尚未在任何生物体中发现。研究基因组结构在抗原变异中的作用的主要障碍之一是抗原基因阵列的高度重复性和杂合性,这阻碍了许多病原体的完整基因组组装。在这里,我们报告了模型原生动物寄生虫布氏锥虫的长抗原基因阵列的从头单倍型特异性组装和支架,使用长读测序技术和染色体折叠的保守特征4。全基因组染色体构象捕获 (Hi-C) 揭示了基因组的不同分区,抗原编码亚端粒区域折叠成不同的、高度紧凑的隔室。此外,我们还进行了一系列分析——Hi-C、荧光原位杂交、使用测序和单细胞 RNA 测序对转座酶可及的染色质进行分析——这表明组蛋白变体 H3.V 和 H4.V 的缺失增加了抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换, 通过同源重组。我们的分析将组蛋白变体鉴定为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并结合缺失实验,证明组蛋白变体作为基因组结构、染色质构象和抗原变异之间的分子联系。V 通过同源重组增加抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换。我们的分析将组蛋白变异识别为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并与缺失实验相结合,证明组蛋白变异可作为基因组结构、染色质构象和抗原变异之间的分子联系。V 通过同源重组增加抗原基因聚类、跨抗原表达位点的 DNA 可及性和表达的抗原同种型的转换。我们的分析将组蛋白变体鉴定为全局基因组结构、局部染色质构象和抗原变异之间的分子联系。长读长测序允许在布氏锥虫中组装抗原基因阵列,并结合缺失实验,证明组蛋白变体作为基因组结构、染色质构象和抗原变异之间的分子联系。


























 京公网安备 11010802027423号
京公网安备 11010802027423号