Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanistic study of the ligand controlled regioselectivity in iridium catalyzed C–H borylation of aromatic imines†
RSC Advances ( IF 3.9 ) Pub Date : 2018-10-16 00:00:00 , DOI: 10.1039/c8ra07886f Yuhua Liu 1 , Jipei Chen 1 , Kangsheng Zhan 1 , Yiqiang Shen 1 , Hui Gao 2, 3 , Lingmin Yao 1
RSC Advances ( IF 3.9 ) Pub Date : 2018-10-16 00:00:00 , DOI: 10.1039/c8ra07886f Yuhua Liu 1 , Jipei Chen 1 , Kangsheng Zhan 1 , Yiqiang Shen 1 , Hui Gao 2, 3 , Lingmin Yao 1
Affiliation
|
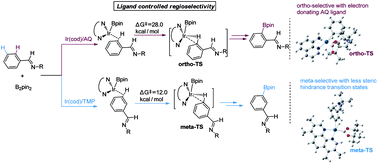
As a major challenge in C–H borylation, how to control the selectivity has attracted lots of attention, however, the related mechanistic information still needs to be uncovered. Herein, density functional theory (DFT) has been used to study the mechanism for the ligand controlled regioselectivity in the iridium-catalyzed C–H borylation of aromatic imines, which is inspired by experimental observations (R. Bisht, B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87). The proposed Ir(I)–Ir(III) catalytic cycle includes (i) the oxidative addition of the C–H bond to iridium(I); (ii) the reductive elimination of a C–B bond; (iii) the oxidative addition of B2pin2 to an iridium(I) hydride complex; and (iv) the reductive elimination of a B–H bond. The oxidative addition of a C–H bond to the iridium center is the determining step. For the ligand AQ, ortho-selectivity is proposed to be attributed to the decreased steric hindrance and increased electron donating effect of AQ (8-aminoquinoline) which promotes proton-transfer in the ortho-transition state of C–H activation. While, for the TMP ligand, the steric repulsion between the TMP (4,5,7,8-tetramethyl-1, 10-phenanthroline) ligand and the ortho-substituted imine hinders the ortho C–H activation and favors meta borylation. Our calculations provide insights into further ligand design to achieve different regioselective borylation of aromatics. Guided by the results, the regioselectivity in the borylation of aromatics may be achieved by accordingly modifying the electronic and steric substituents of the ligand.
中文翻译:

铱催化芳族亚胺 C-H 硼化反应中配体控制区域选择性的机理研究†
作为 C-H 硼化的主要挑战,如何控制选择性受到了广泛关注,但相关的机理信息仍有待挖掘。在此,密度泛函理论 (DFT) 已被用于研究铱催化芳族亚胺 C-H 硼化反应中配体控制的区域选择性机制,其灵感来自实验观察 (R. Bisht, B. Chattopadhyay, J.美国化学学会, 2016, 138 , 84–87)。提议的 Ir( I )-Ir( III ) 催化循环包括 (i) C-H 键与铱( I ) 的氧化加成;(ii) C-B 键的还原消除;(iii) B 2 pin 2的氧化加成为氢化铱 ( I ) 络合物;(iv) B-H 键的还原消除。铱中心的 C-H 键的氧化加成是决定性步骤。对于配体 AQ,邻位选择性被认为是由于 AQ(8-氨基喹啉)的空间位阻降低和给电子效应增加,这促进了 C-H 活化的邻位过渡态的质子转移。而对于 TMP 配体,TMP (4,5,7,8-tetramethyl-1, 10-phenanthroline) 配体与邻位取代的亚胺之间的空间排斥阻碍了邻位C-H 活化并有利于间位硼化。我们的计算为进一步的配体设计提供了见解,以实现芳烃的不同区域选择性硼化。在结果的指导下,可以通过相应地修饰配体的电子和空间取代基来实现芳烃硼基化的区域选择性。
更新日期:2018-10-16
中文翻译:
铱催化芳族亚胺 C-H 硼化反应中配体控制区域选择性的机理研究†
作为 C-H 硼化的主要挑战,如何控制选择性受到了广泛关注,但相关的机理信息仍有待挖掘。在此,密度泛函理论 (DFT) 已被用于研究铱催化芳族亚胺 C-H 硼化反应中配体控制的区域选择性机制,其灵感来自实验观察 (R. Bisht, B. Chattopadhyay, J.美国化学学会, 2016, 138 , 84–87)。提议的 Ir( I )-Ir( III ) 催化循环包括 (i) C-H 键与铱( I ) 的氧化加成;(ii) C-B 键的还原消除;(iii) B 2 pin 2的氧化加成为氢化铱 ( I ) 络合物;(iv) B-H 键的还原消除。铱中心的 C-H 键的氧化加成是决定性步骤。对于配体 AQ,邻位选择性被认为是由于 AQ(8-氨基喹啉)的空间位阻降低和给电子效应增加,这促进了 C-H 活化的邻位过渡态的质子转移。而对于 TMP 配体,TMP (4,5,7,8-tetramethyl-1, 10-phenanthroline) 配体与邻位取代的亚胺之间的空间排斥阻碍了邻位C-H 活化并有利于间位硼化。我们的计算为进一步的配体设计提供了见解,以实现芳烃的不同区域选择性硼化。在结果的指导下,可以通过相应地修饰配体的电子和空间取代基来实现芳烃硼基化的区域选择性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号