当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Joint Precursor Elution Profile Inference via Regression for Peptide Detection in Data-Independent Acquisition Mass Spectra.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-26 , DOI: 10.1021/acs.jproteome.8b00365 Alex Hu , Yang Young Lu , Jeff Bilmes , William Stafford Noble
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-26 , DOI: 10.1021/acs.jproteome.8b00365 Alex Hu , Yang Young Lu , Jeff Bilmes , William Stafford Noble
|
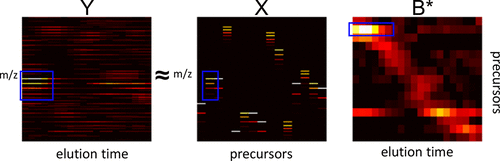
In data independent acquisition (DIA) mass spectrometry, precursor scans are interleaved with wide-window fragmentation scans, resulting in complex fragmentation spectra containing multiple coeluting peptide species. In this setting, detecting the isotope distribution profiles of intact peptides in the precursor scans can be a critical initial step in accurate peptide detection and quantification. This peak detection step is particularly challenging when the isotope peaks associated with two different peptide species overlap-or interfere-with one another. We propose a regression model, called Siren, to detect isotopic peaks in precursor DIA data that can explicitly account for interference. We validate Siren's peak-calling performance on a variety of data sets by counting how many of the peaks Siren identifies are associated with confidently detected peptides. In particular, we demonstrate that substituting the Siren regression model in place of the existing peak-calling step in DIA-Umpire leads to improved overall rates of peptide detection.
中文翻译:

通过回归进行联合前体洗脱曲线推断,用于数据独立的采集质谱中的肽检测。
在数据独立采集 (DIA) 质谱中,前体扫描与宽窗口碎片扫描交织在一起,产生包含多种共洗脱肽种类的复杂碎片光谱。在这种情况下,检测前体扫描中完整肽的同位素分布图可能是准确肽检测和定量的关键初始步骤。当与两种不同肽种类相关的同位素峰相互重叠或干扰时,这一峰检测步骤尤其具有挑战性。我们提出了一种称为 Siren 的回归模型,用于检测前体 DIA 数据中的同位素峰,该数据可以明确解释干扰。我们通过计算 Siren 识别的峰中有多少与可靠检测到的肽相关,来验证 Siren 在各种数据集上的峰识别性能。特别是,我们证明用 Siren 回归模型代替 DIA-Umpire 中现有的峰值调用步骤可以提高肽检测的总体率。
更新日期:2018-10-27
中文翻译:
通过回归进行联合前体洗脱曲线推断,用于数据独立的采集质谱中的肽检测。
在数据独立采集 (DIA) 质谱中,前体扫描与宽窗口碎片扫描交织在一起,产生包含多种共洗脱肽种类的复杂碎片光谱。在这种情况下,检测前体扫描中完整肽的同位素分布图可能是准确肽检测和定量的关键初始步骤。当与两种不同肽种类相关的同位素峰相互重叠或干扰时,这一峰检测步骤尤其具有挑战性。我们提出了一种称为 Siren 的回归模型,用于检测前体 DIA 数据中的同位素峰,该数据可以明确解释干扰。我们通过计算 Siren 识别的峰中有多少与可靠检测到的肽相关,来验证 Siren 在各种数据集上的峰识别性能。特别是,我们证明用 Siren 回归模型代替 DIA-Umpire 中现有的峰值调用步骤可以提高肽检测的总体率。










































 京公网安备 11010802027423号
京公网安备 11010802027423号