当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
XINA: A Workflow for the Integration of Multiplexed Proteomics Kinetics Data with Network Analysis.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-29 , DOI: 10.1021/acs.jproteome.8b00615 Lang Ho Lee 1 , Arda Halu 1, 2 , Stephanie Morgan 1 , Hiroshi Iwata 1 , Masanori Aikawa 1, 2, 3 , Sasha A Singh 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-10-29 , DOI: 10.1021/acs.jproteome.8b00615 Lang Ho Lee 1 , Arda Halu 1, 2 , Stephanie Morgan 1 , Hiroshi Iwata 1 , Masanori Aikawa 1, 2, 3 , Sasha A Singh 1
Affiliation
|
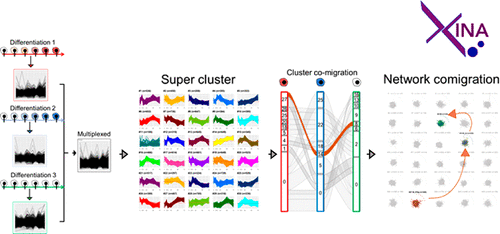
Quantitative proteomics experiments, using for instance isobaric tandem mass tagging approaches, are conducive to measuring changes in protein abundance over multiple time points in response to one or more conditions or stimulations. The aim is often to determine which proteins exhibit similar patterns within and across experimental conditions, since proteins with coabundance patterns may have common molecular functions related to a given stimulation. In order to facilitate the identification and analyses of coabundance patterns within and across conditions, we previously developed a software inspired by the isobaric mass tagging method itself. Specifically, multiple data sets are tagged in silico and combined for subsequent subgrouping into multiple clusters within a single output depicting the variation across all conditions, converting a typical inter-data-set comparison into an intra-data-set comparison. An updated version of our software, XINA, not only extracts coabundance profiles within and across experiments but also incorporates protein-protein interaction databases and integrative resources such as KEGG to infer interactors and molecular functions, respectively, and produces intuitive graphical outputs. In this report, we compare the kinetics profiles of >5600 unique proteins derived from three macrophage cell culture experiments and demonstrate through intuitive visualizations that XINA identifies key regulators of macrophage activation via their coabundance patterns.
中文翻译:

XINA:多重蛋白质组动力学数据与网络分析集成的工作流程。
定量蛋白质组学实验,例如使用同量异位串联质量标记方法,有利于测量响应一种或多种条件或刺激的多个时间点上蛋白质丰度的变化。目的通常是确定哪些蛋白质在实验条件内和跨实验条件表现出相似的模式,因为具有共丰度模式的蛋白质可能具有与给定刺激相关的共同分子功能。为了促进条件内和跨条件下的共丰度模式的识别和分析,我们之前开发了一款受同量异位质量标记方法本身启发的软件。具体来说,多个数据集在计算机中被标记并组合起来,以便随后在单个输出中分组为多个集群,描述所有条件下的变化,将典型的数据集间比较转换为数据集内比较。我们软件的更新版本 XINA 不仅可以提取实验内和实验间的共丰度曲线,还结合了蛋白质-蛋白质相互作用数据库和 KEGG 等综合资源,分别推断相互作用因子和分子功能,并生成直观的图形输出。在本报告中,我们比较了来自三个巨噬细胞培养实验的超过 5600 种独特蛋白质的动力学特征,并通过直观的可视化证明 XINA 通过其共丰度模式识别了巨噬细胞激活的关键调节因子。
更新日期:2018-10-30
中文翻译:
XINA:多重蛋白质组动力学数据与网络分析集成的工作流程。
定量蛋白质组学实验,例如使用同量异位串联质量标记方法,有利于测量响应一种或多种条件或刺激的多个时间点上蛋白质丰度的变化。目的通常是确定哪些蛋白质在实验条件内和跨实验条件表现出相似的模式,因为具有共丰度模式的蛋白质可能具有与给定刺激相关的共同分子功能。为了促进条件内和跨条件下的共丰度模式的识别和分析,我们之前开发了一款受同量异位质量标记方法本身启发的软件。具体来说,多个数据集在计算机中被标记并组合起来,以便随后在单个输出中分组为多个集群,描述所有条件下的变化,将典型的数据集间比较转换为数据集内比较。我们软件的更新版本 XINA 不仅可以提取实验内和实验间的共丰度曲线,还结合了蛋白质-蛋白质相互作用数据库和 KEGG 等综合资源,分别推断相互作用因子和分子功能,并生成直观的图形输出。在本报告中,我们比较了来自三个巨噬细胞培养实验的超过 5600 种独特蛋白质的动力学特征,并通过直观的可视化证明 XINA 通过其共丰度模式识别了巨噬细胞激活的关键调节因子。










































 京公网安备 11010802027423号
京公网安备 11010802027423号