当前位置:
X-MOL 学术
›
J. Mater. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The competition between mechanical stability and charge carrier mobility in MA-based hybrid perovskites: insight from DFT†
Journal of Materials Chemistry C ( IF 5.7 ) Pub Date : 2018-10-08 00:00:00 , DOI: 10.1039/c8tc04750b Jung-Hoon Lee 1, 2, 3, 4 , Zeyu Deng 1, 2, 3, 4 , Nicholas C. Bristowe 4, 5, 6, 7, 8 , Paul D. Bristowe 1, 2, 3, 4 , Anthony K. Cheetham 1, 2, 3, 4
Journal of Materials Chemistry C ( IF 5.7 ) Pub Date : 2018-10-08 00:00:00 , DOI: 10.1039/c8tc04750b Jung-Hoon Lee 1, 2, 3, 4 , Zeyu Deng 1, 2, 3, 4 , Nicholas C. Bristowe 4, 5, 6, 7, 8 , Paul D. Bristowe 1, 2, 3, 4 , Anthony K. Cheetham 1, 2, 3, 4
Affiliation
|
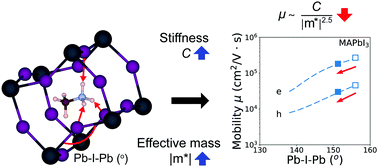
Hybrid organic–inorganic perovskites and their inorganic analogues, such as MAPbI3 (MA = methylammonium, CH3NH3) and CsPbI3, are currently under intense investigation due to their high-power conversion efficiencies and low cost for solar cell applications. Herein, we investigate the effect of methylammonium and the inorganic A-cations on the elastic and related transport properties of halide perovskites using van der Waals (vdW) corrected density functional theory (DFT) calculations. For inorganic halide perovskites we find that the bonding within the inorganic framework is mainly responsible for their elastic behavior. However, our DFT calculations show that when a MA cation is substituted into the structure the combined effects of stericity (conformation) and hydrogen-framework interactions improve the material's resistance to deformation. For example, the orientationally-averaged Young's modulus of orthorhombic MAPbI3 increases by about 19% compared to the equivalent inorganic series of structures. We also find that, within the carrier-acoustic phonon scattering regime, the electron and hole carrier mobilities of hybrid halide perovskites are lowered by the hydrogen-bonding-induced tilting of the inorganic octahedra. Taken together, these results can help guide the optimization of the mechanical and transport properties of perovskite-based solar cell materials.
中文翻译:

基于MA的混合钙钛矿中机械稳定性和电荷载流子迁移率之间的竞争:DFT的见解†
杂化的有机-无机钙钛矿及其无机类似物,例如MAPbI 3(MA =甲基铵,CH 3 NH 3)和CsPbI 3由于它们的高功率转换效率和太阳能电池应用的低成本,目前正受到广泛研究。本文中,我们使用范德华(vdW)校正的密度泛函理论(DFT)计算研究了甲基铵和无机A-阳离子对卤化物钙钛矿的弹性和相关运输性质的影响。对于无机卤化物钙钛矿,我们发现无机骨架内的键合主要负责其弹性行为。但是,我们的DFT计算表明,当MA阳离子被取代到结构中时,空间性(构象)和氢-骨架相互作用的综合作用提高了材料的抗变形能力。例如,正交MAPbI 3的取向平均杨氏模量与同等的无机系列结构相比,它增加了约19%。我们还发现,在载流子-声子声子散射范围内,杂化卤化物钙钛矿的电子和空穴载流子迁移率由于氢键诱导的无机八面体的倾斜而降低。综上所述,这些结果可以帮助指导钙钛矿基太阳能电池材料的机械性能和运输性能的优化。
更新日期:2018-10-08
中文翻译:
基于MA的混合钙钛矿中机械稳定性和电荷载流子迁移率之间的竞争:DFT的见解†
杂化的有机-无机钙钛矿及其无机类似物,例如MAPbI 3(MA =甲基铵,CH 3 NH 3)和CsPbI 3由于它们的高功率转换效率和太阳能电池应用的低成本,目前正受到广泛研究。本文中,我们使用范德华(vdW)校正的密度泛函理论(DFT)计算研究了甲基铵和无机A-阳离子对卤化物钙钛矿的弹性和相关运输性质的影响。对于无机卤化物钙钛矿,我们发现无机骨架内的键合主要负责其弹性行为。但是,我们的DFT计算表明,当MA阳离子被取代到结构中时,空间性(构象)和氢-骨架相互作用的综合作用提高了材料的抗变形能力。例如,正交MAPbI 3的取向平均杨氏模量与同等的无机系列结构相比,它增加了约19%。我们还发现,在载流子-声子声子散射范围内,杂化卤化物钙钛矿的电子和空穴载流子迁移率由于氢键诱导的无机八面体的倾斜而降低。综上所述,这些结果可以帮助指导钙钛矿基太阳能电池材料的机械性能和运输性能的优化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号