当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Best of Two Worlds? How MD Simulations of Amphiphilic Helical Peptides in Membranes Can Complement Data from Oriented Solid-State NMR
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-10-05 00:00:00 , DOI: 10.1021/acs.jctc.8b00283 Sabine Reißer 1 , Erik Strandberg 2 , Thomas Steinbrecher 3 , Marcus Elstner 3 , Anne S. Ulrich 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-10-05 00:00:00 , DOI: 10.1021/acs.jctc.8b00283 Sabine Reißer 1 , Erik Strandberg 2 , Thomas Steinbrecher 3 , Marcus Elstner 3 , Anne S. Ulrich 1, 2
Affiliation
|
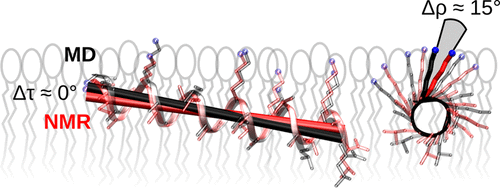
The membrane alignment of helical amphiphilic peptides in oriented phospholipid bilayers can be obtained as ensemble and time averages from solid state 2H NMR by fitting the quadrupolar splittings to ideal α-helices. At the same time, molecular dynamics (MD) simulations can provide atomistic insight into peptide–membrane systems. Here, we evaluate the potential of MD simulations to complement the experimental NMR data that is available on three exemplary systems: the natural antimicrobial peptide PGLa and the two designer-made peptides MSI-103 and KIA14, whose sequences were derived from PGLa. Each peptide was simulated for 1 μs in a DMPC lipid bilayer. We calculated from the MD simulations the local angles which define the side chain geometry with respect to the peptide helix. The peptide orientation was then calculated (i) directly from the simulation, (ii) from back-calculated MD-derived NMR splittings, and (iii) from experimental 2H NMR splittings. Our findings are that (1) the membrane orientation and secondary structure of the peptides found in the NMR analysis are generally well reproduced by the simulations; (2) the geometry of the side chains with respect to the helix backbone can deviate significantly from the ideal structure depending on the specific residue, but on average all side chains have the same orientation; and (3) for all of our peptides, the azimuthal rotation angle found from the MD-derived splittings is about 15° smaller than the experimental value.
中文翻译:

最好的两个世界?膜中两亲性螺旋肽的MD模拟如何补充来自定向固态NMR的数据
取向的磷脂双层膜中螺旋两亲性肽的膜比对可从固态2获得的集合和时间平均值获得通过将四极分裂拟合为理想的α螺旋进行1 H NMR。同时,分子动力学(MD)模拟可以提供对肽-膜系统的原子学见解。在这里,我们评估MD模拟的潜力,以补充在三个示例性系统上可获得的实验NMR数据:天然抗菌肽PGLa和两个设计制造的肽MSI-103和KIA14,其序列均来自PGLa。在DMPC脂质双层中模拟每种肽1 s。我们从MD模拟中计算出局部角度,这些局部角度定义了相对于肽螺旋的侧链几何形状。然后(i)直接从模拟中计算出肽的方向,(ii)从反向计算的MD衍生的NMR拆分中计算出,并且(iii)从实验2中计算出1 H NMR分裂。我们的发现是:(1)在NMR分析中发现的肽的膜取向和二级结构通常可以通过模拟得到很好的再现;(2)取决于特定的残基,相对于螺旋骨架的侧链的几何形状可以明显偏离理想结构,但是平均而言,所有侧链具有相同的取向;(3)对于我们所有的肽,从MD衍生的片段中发现的方位角旋转角均比实验值小15°。
更新日期:2018-10-05
中文翻译:
最好的两个世界?膜中两亲性螺旋肽的MD模拟如何补充来自定向固态NMR的数据
取向的磷脂双层膜中螺旋两亲性肽的膜比对可从固态2获得的集合和时间平均值获得通过将四极分裂拟合为理想的α螺旋进行1 H NMR。同时,分子动力学(MD)模拟可以提供对肽-膜系统的原子学见解。在这里,我们评估MD模拟的潜力,以补充在三个示例性系统上可获得的实验NMR数据:天然抗菌肽PGLa和两个设计制造的肽MSI-103和KIA14,其序列均来自PGLa。在DMPC脂质双层中模拟每种肽1 s。我们从MD模拟中计算出局部角度,这些局部角度定义了相对于肽螺旋的侧链几何形状。然后(i)直接从模拟中计算出肽的方向,(ii)从反向计算的MD衍生的NMR拆分中计算出,并且(iii)从实验2中计算出1 H NMR分裂。我们的发现是:(1)在NMR分析中发现的肽的膜取向和二级结构通常可以通过模拟得到很好的再现;(2)取决于特定的残基,相对于螺旋骨架的侧链的几何形状可以明显偏离理想结构,但是平均而言,所有侧链具有相同的取向;(3)对于我们所有的肽,从MD衍生的片段中发现的方位角旋转角均比实验值小15°。










































 京公网安备 11010802027423号
京公网安备 11010802027423号