当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Discerning Chemical Pressure amidst Weak Potentials: Vibrational Modes and Dumbbell/Atom Substitution in Intermetallic Aluminides
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-10-02 00:00:00 , DOI: 10.1021/acs.jpca.8b07419 Katerina P. Hilleke 1 , Daniel C. Fredrickson 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-10-02 00:00:00 , DOI: 10.1021/acs.jpca.8b07419 Katerina P. Hilleke 1 , Daniel C. Fredrickson 1
Affiliation
|
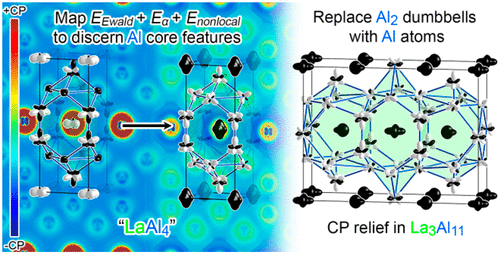
The space requirements of atoms are generally regarded as key constraints in the structures, reactivity, and physical properties of chemical systems. However, the empirical nature of such considerations renders the elucidation of these size effects with first-principles calculations challenging. DFT-chemical pressure (DFT-CP) analysis, in which the output of DFT calculations is used to construct maps of the local pressures acting between atoms due to lattice constraints, is one productive approach to extracting the role of atomic size in the crystal structures of materials. While in principle this method should be applicable to any system for which DFT is deemed an appropriate treatment, so far it has worked most successfully when semicore electrons are included in the valence set of each atom to supply an explicit repulsive response to compression. In this Article, we address this limiting factor, using as model systems intermetallics based on aluminum, a key component in many structurally interesting phases that is not amenable to modeling with a semicore pseudopotential. Beginning with the Laves phase CaAl2, we illustrate the difficulties of creating a CP scheme that reflects the compound’s phonon band structure with the original method due to minimal core responses on the Al atoms. These deficiencies are resolved through a spatial mapping of three energetic terms that were previously treated as homogeneous background effects: the Ewald, Eα, and nonlocal pseudopotential components. When charge transfer is factored into the integration scheme, CP schemes consistent with the phonon band structure are obtainable for CaAl2, regardless of whether Ca is modeled with a semicore or valence-only pseudopotential. Finally, we demonstrate the utility of the revised method through its application to the La3Al11 structure, which is shown to soothe CPs that would be encountered in a hypothetical BaAl4-type parent phase through the substitution of selected Al2 pairs with single Al atoms. La3Al11 then emerges as an example of a more general phenomenon, CP-driven substitutions of simple motifs.
中文翻译:

在弱电势中识别化学压力:金属间化合物铝化物中的振动模式和哑铃/原子取代
原子的空间要求通常被视为化学系统结构,反应性和物理性质的关键限制。然而,这些考虑因素的经验性质使得对这些尺寸效应的阐明与第一性原理的计算具有挑战性。DFT-化学压力(DFT-CP)分析(其中DFT计算的输出用于构建由于晶格约束而作用于原子之间的局部压力的图)是一种提取原子尺寸在晶体结构中的作用的有效方法材料。虽然原则上该方法应适用于DFT被认为是适当处理方法的任何系统,到目前为止,当每个原子的化合价中包含半核电子以提供对压缩的显式排斥响应时,该方法最为成功。在本文中,我们通过使用基于铝的金属间化合物作为模型系统来解决此限制因素,铝是许多结构上令人感兴趣的阶段中的关键组件,这些阶段不适合使用半芯假电势进行建模。从Laves相CaAl开始如图2所示,由于铝原子上的最小核响应,我们说明了用原始方法创建能反映化合物声子能带结构的CP方案的困难。的埃瓦尔德,:这些缺陷是通过三个能量方面的空间映射先前为均匀的背景效果处理解决ë α,和非局部赝组件。当将电荷转移计入积分方案时,对于CaAl 2可以获得与声子能带结构一致的CP方案,而不管Ca是用半核还是仅化合价的伪势建模的。最后,我们通过将其应用于La 3 Al 11来演示该修订方法的实用性结构表明,通过选择的Al 2对替换为单个Al原子,可以缓解在假设的BaAl 4型母相中可能遇到的CP 。然后,La 3 Al 11作为更普遍的现象的一个例子出现,即CP驱动的简单图案的替代。
更新日期:2018-10-02
中文翻译:
在弱电势中识别化学压力:金属间化合物铝化物中的振动模式和哑铃/原子取代
原子的空间要求通常被视为化学系统结构,反应性和物理性质的关键限制。然而,这些考虑因素的经验性质使得对这些尺寸效应的阐明与第一性原理的计算具有挑战性。DFT-化学压力(DFT-CP)分析(其中DFT计算的输出用于构建由于晶格约束而作用于原子之间的局部压力的图)是一种提取原子尺寸在晶体结构中的作用的有效方法材料。虽然原则上该方法应适用于DFT被认为是适当处理方法的任何系统,到目前为止,当每个原子的化合价中包含半核电子以提供对压缩的显式排斥响应时,该方法最为成功。在本文中,我们通过使用基于铝的金属间化合物作为模型系统来解决此限制因素,铝是许多结构上令人感兴趣的阶段中的关键组件,这些阶段不适合使用半芯假电势进行建模。从Laves相CaAl开始如图2所示,由于铝原子上的最小核响应,我们说明了用原始方法创建能反映化合物声子能带结构的CP方案的困难。的埃瓦尔德,:这些缺陷是通过三个能量方面的空间映射先前为均匀的背景效果处理解决ë α,和非局部赝组件。当将电荷转移计入积分方案时,对于CaAl 2可以获得与声子能带结构一致的CP方案,而不管Ca是用半核还是仅化合价的伪势建模的。最后,我们通过将其应用于La 3 Al 11来演示该修订方法的实用性结构表明,通过选择的Al 2对替换为单个Al原子,可以缓解在假设的BaAl 4型母相中可能遇到的CP 。然后,La 3 Al 11作为更普遍的现象的一个例子出现,即CP驱动的简单图案的替代。










































 京公网安备 11010802027423号
京公网安备 11010802027423号