Nature Reviews Chemistry ( IF 38.1 ) Pub Date : 2018-10-01 , DOI: 10.1038/s41570-018-0040-8 Jolene P. Reid , Matthew S. Sigman
|
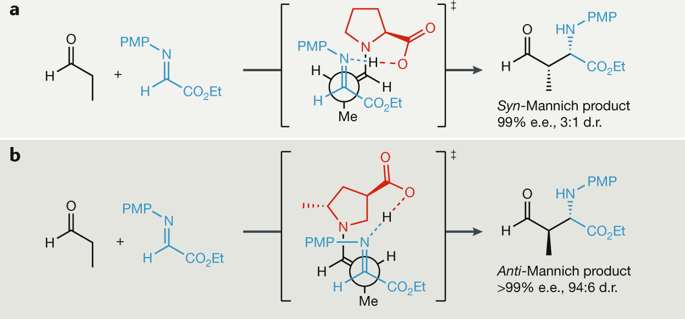
Advances in density functional theory (DFT) mean that it is now possible to study catalytic reactions with sufficient accuracy that the results compare favourably with experiment. These high-level calculations have been applied to understand and predict variations in catalytic performance from one catalyst to another, but can require substantial computational resources. By contrast, multivariate linear regression (MLR) methods are rapidly becoming versatile, statistical tools for predicting and understanding the roles of catalysts and substrates and act as a useful complement to complex transition state calculations, with a substantially lower computational cost. Herein, we compare these approaches, DFT calculations and data analysis techniques, and discuss their ability to provide meaningful predictions of catalyst performance. Examples of applications are selected to demonstrate the advantages and limitations of both tools. Several ongoing challenges in the predictions of reaction outcomes are also highlighted.
中文翻译:
比较发现小分子手性催化剂的定量预测方法
密度泛函理论(DFT)的发展意味着现在有可能以足够的精度研究催化反应,从而使结果与实验相吻合。这些高级计算已用于理解和预测从一种催化剂到另一种催化剂的催化性能变化,但可能需要大量的计算资源。相比之下,多元线性回归(MLR)方法正迅速成为通用的统计工具,用于预测和理解催化剂和底物的作用,并以显着较低的计算成本作为复杂过渡态计算的有用补充。在本文中,我们比较了这些方法,DFT计算和数据分析技术,并讨论了它们提供有意义的催化剂性能预测的能力。选择应用程序示例来演示这两种工具的优点和局限性。在反应结果的预测中还突出了一些正在进行的挑战。










































 京公网安备 11010802027423号
京公网安备 11010802027423号