当前位置:
X-MOL 学术
›
ACS Earth Space Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantum Chemical Approach for Calculating Stability Constants of Mercury Complexes
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2018-09-28 00:00:00 , DOI: 10.1021/acsearthspacechem.8b00102 Deepa Devarajan 1, 2 , Peng Lian 1 , Scott C. Brooks 3 , Jerry M. Parks 1 , Jeremy C. Smith 1, 2
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2018-09-28 00:00:00 , DOI: 10.1021/acsearthspacechem.8b00102 Deepa Devarajan 1, 2 , Peng Lian 1 , Scott C. Brooks 3 , Jerry M. Parks 1 , Jeremy C. Smith 1, 2
Affiliation
|
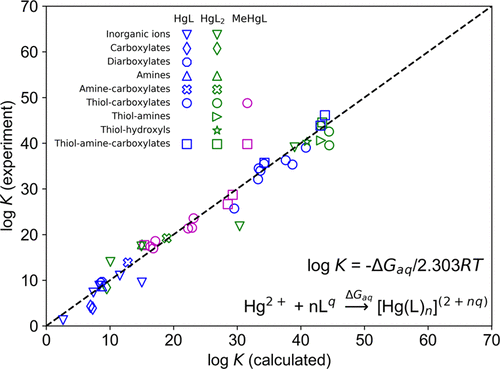
Stability constants are central to the multiscale modeling of the thermodynamic speciation, cycling, and transport of mercury (Hg) and other contaminants in aquatic environments. However, for Hg, experimental values for many relevant complexes are not available, and for others can span ranges in excess of 10 log units. The missing data and large uncertainties lead to significant knowledge gaps in predictions of thermodynamic speciation. As an alternative to experimental measurements, thermodynamic quantities can be calculated with quantum chemical methods. Among these, density functional theory (DFT) with a polarizable continuum solvent combines accuracy with practicability. Here, we present an accurate and quick approach in which we use DFT with continuum solvation to calculate stability constants of Hg complexes with inorganic and low molecular-weight organic ligands in aqueous solution. Specifically, we use the M06/[SDD]6-31+G(d,p) level of theory in combination with a modified version of the SMD solvent model in which the solute radii are reoptimized with a scaled solvent-accessible surface approach. For the set of 37 Hg complexes used for optimization, which contain environmentally relevant functional groups and have reliable experimental stability constants, we obtain a mean unsigned error of 1.4 log units. Testing the method on an independent set of 12 Hg complexes reproduces the experimental stability constants to a mean unsigned error of 1.6 log units. This approach is a substantial step toward generally applicable rapid stability constant derivation for a wide range of Hg complexes, including those present in dissolved organic matter.
中文翻译:

汞络合物稳定常数的量子化学方法
稳定性常数对于水生环境中汞(Hg)和其他污染物的热力学形态,循环和运输的多尺度建模至关重要。但是,对于汞,许多相关配合物的实验值不可用,而对于其他汞,其实验值范围可能超过10个对数单位。缺少的数据和较大的不确定性导致热力学形态预测中的重大知识空白。作为实验测量的替代方法,可以使用量子化学方法来计算热力学量。其中,使用可极化连续介质溶剂的密度泛函理论(DFT)将准确性与实用性结合在一起。这里,我们提出了一种准确而快速的方法,其中我们将DFT与连续溶剂化一起使用,以计算具有无机和低分子量有机配体的Hg配合物在水溶液中的稳定常数。具体来说,我们将M06 / [SDD] 6-31 + G(d,p)的理论水平与SMD溶剂模型的修改版本结合使用,在该模型中,溶质半径通过可缩放的溶剂可及表面方法进行了重新优化。对于用于优化的37个Hg配合物集,这些配合物包含与环境相关的官能团并具有可靠的实验稳定性常数,我们获得的平均无符号误差为1.4 log单位。在独立的12个Hg配合物上测试该方法可将实验稳定性常数重现为1.6 log个单位的平均无符号误差。
更新日期:2018-09-28
中文翻译:
汞络合物稳定常数的量子化学方法
稳定性常数对于水生环境中汞(Hg)和其他污染物的热力学形态,循环和运输的多尺度建模至关重要。但是,对于汞,许多相关配合物的实验值不可用,而对于其他汞,其实验值范围可能超过10个对数单位。缺少的数据和较大的不确定性导致热力学形态预测中的重大知识空白。作为实验测量的替代方法,可以使用量子化学方法来计算热力学量。其中,使用可极化连续介质溶剂的密度泛函理论(DFT)将准确性与实用性结合在一起。这里,我们提出了一种准确而快速的方法,其中我们将DFT与连续溶剂化一起使用,以计算具有无机和低分子量有机配体的Hg配合物在水溶液中的稳定常数。具体来说,我们将M06 / [SDD] 6-31 + G(d,p)的理论水平与SMD溶剂模型的修改版本结合使用,在该模型中,溶质半径通过可缩放的溶剂可及表面方法进行了重新优化。对于用于优化的37个Hg配合物集,这些配合物包含与环境相关的官能团并具有可靠的实验稳定性常数,我们获得的平均无符号误差为1.4 log单位。在独立的12个Hg配合物上测试该方法可将实验稳定性常数重现为1.6 log个单位的平均无符号误差。









































 京公网安备 11010802027423号
京公网安备 11010802027423号