当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Study of Protein–Ligand Interactions Using the Molecules-in-Molecules Fragmentation-Based Method
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-28 00:00:00 , DOI: 10.1021/acs.jctc.8b00531 Bishnu Thapa 1 , Daniel Beckett 1 , Jon Erickson 2 , Krishnan Raghavachari 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-28 00:00:00 , DOI: 10.1021/acs.jctc.8b00531 Bishnu Thapa 1 , Daniel Beckett 1 , Jon Erickson 2 , Krishnan Raghavachari 1
Affiliation
|
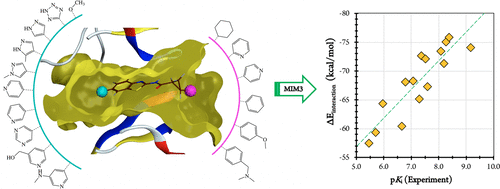
We have recently significantly expanded the applicability of our Molecules-in-Molecules (MIM) fragmentation method to large proteins by developing a three-layer model (MIM3) in which an accurate quantum-mechanical method is used in conjunction with a cost-effective, dispersion-corrected semiempirical model to overcome previous computational bottlenecks. In this work, we develop MIM3 as a structure-based drug design tool by application of the methodology for the accurate calculation of protein–ligand interaction energies. A systematic protocol is derived for the determination of the geometries of the protein–ligand complexes and to calculate their accurate interaction energies in the gas phase using MIM3. We also derive a simple and affordable procedure based on implicit solvation models and the ligand solvent-accessible surface area to approximate the ligand desolvation penalty in gas-phase interaction energy calculations. We have carefully assessed how closely such interaction energies, which are based on a single protein–ligand conformation, display correlations with the experimentally determined binding affinities. The performance of MIM3 was evaluated on a total of seven data sets comprising 89 protein–ligand complexes, all with experimentally known binding affinities, using a binding pocket involving a quantum region ranging in size from 250 to 600 atoms. The dispersion-corrected B97-D3BJ density functional, previously known to perform accurately for calculations involving non-covalent interactions, was used as the target level of theory for this work, with dispersion-corrected PM6-D3 as the semiempirical low level to incorporate the long-range interactions. Comparing directly to the experimental binding potencies, we obtain impressive correlations over all seven test sets, with an R2 range of 0.74–0.93 and a Spearman rank correlation coefficient (ρ) range of 0.83–0.93. Our results suggest that protein–ligand interaction energies are useful in predicting binding potency trends and validate the potential of MIM3 as a quantum-chemical structure-based drug design tool.
中文翻译:

基于分子中分子碎片化方法的蛋白质-配体相互作用的理论研究
我们最近开发了三层模型(MIM3),在其中将精确的量子力学方法与具有成本效益的方法结合使用,从而大大扩展了分子内分子(MIM)片段化方法对大蛋白的适用性,色散校正的半经验模型可以克服以前的计算瓶颈。在这项工作中,我们通过应用可精确计算蛋白质-配体相互作用能的方法,将MIM3开发为基于结构的药物设计工具。导出了用于确定蛋白质-配体复合物的几何形状并使用MIM3在气相中计算其精确相互作用能的系统协议。我们还基于隐式溶剂化模型和配体溶剂可及的表面积推导了一种简单且可负担的程序,以估算气相相互作用能计算中的配体去溶剂化损失。我们已经仔细评估了基于单一蛋白质-配体构象的这种相互作用能显示出与实验确定的结合亲和力之间的相关性。使用包含大小范围从250到600个原子的结合袋,对包括89个蛋白-配体复合物(均具有实验已知的结合亲和力)的总共7个数据集进行了评估,以评估MIM3的性能。经过分散校正的B97-D3BJ密度泛函,以前已知能够精确地执行涉及非共价相互作用的计算,作为这项工作的理论目标水平,使用色散校正的PM6-D3作为结合长期相互作用的半经验性低水平。直接与实验结合力进行比较,我们在所有七个测试集上获得了令人印象深刻的相关性,R 2范围为0.74-0.93,Spearman等级相关系数(ρ)范围为0.83-0.93。我们的结果表明,蛋白质-配体相互作用能可用于预测结合力趋势,并验证MIM3作为基于量子化学结构的药物设计工具的潜力。
更新日期:2018-09-28
中文翻译:
基于分子中分子碎片化方法的蛋白质-配体相互作用的理论研究
我们最近开发了三层模型(MIM3),在其中将精确的量子力学方法与具有成本效益的方法结合使用,从而大大扩展了分子内分子(MIM)片段化方法对大蛋白的适用性,色散校正的半经验模型可以克服以前的计算瓶颈。在这项工作中,我们通过应用可精确计算蛋白质-配体相互作用能的方法,将MIM3开发为基于结构的药物设计工具。导出了用于确定蛋白质-配体复合物的几何形状并使用MIM3在气相中计算其精确相互作用能的系统协议。我们还基于隐式溶剂化模型和配体溶剂可及的表面积推导了一种简单且可负担的程序,以估算气相相互作用能计算中的配体去溶剂化损失。我们已经仔细评估了基于单一蛋白质-配体构象的这种相互作用能显示出与实验确定的结合亲和力之间的相关性。使用包含大小范围从250到600个原子的结合袋,对包括89个蛋白-配体复合物(均具有实验已知的结合亲和力)的总共7个数据集进行了评估,以评估MIM3的性能。经过分散校正的B97-D3BJ密度泛函,以前已知能够精确地执行涉及非共价相互作用的计算,作为这项工作的理论目标水平,使用色散校正的PM6-D3作为结合长期相互作用的半经验性低水平。直接与实验结合力进行比较,我们在所有七个测试集上获得了令人印象深刻的相关性,R 2范围为0.74-0.93,Spearman等级相关系数(ρ)范围为0.83-0.93。我们的结果表明,蛋白质-配体相互作用能可用于预测结合力趋势,并验证MIM3作为基于量子化学结构的药物设计工具的潜力。








































 京公网安备 11010802027423号
京公网安备 11010802027423号