当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
t-Distributed Stochastic Neighbor Embedding Method with the Least Information Loss for Macromolecular Simulations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-25 00:00:00 , DOI: 10.1021/acs.jctc.8b00652 Hongyu Zhou 1 , Feng Wang 1 , Peng Tao 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-25 00:00:00 , DOI: 10.1021/acs.jctc.8b00652 Hongyu Zhou 1 , Feng Wang 1 , Peng Tao 1
Affiliation
|
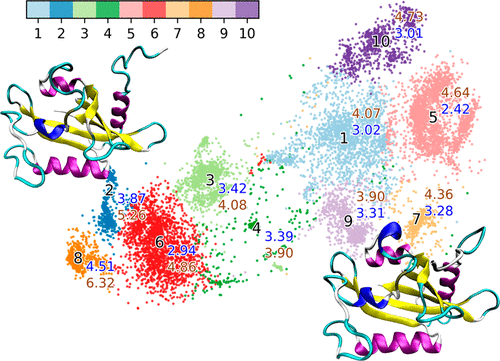
Dimensionality reduction methods are usually applied on molecular dynamics simulations of macromolecules for analysis and visualization purposes. It is normally desired that suitable dimensionality reduction methods could clearly distinguish functionally important states with different conformations for the systems of interest. However, common dimensionality reduction methods for macromolecules simulations, including predefined order parameters and collective variables (CVs), principal component analysis (PCA), and time-structure based independent component analysis (t-ICA), only have limited success due to significant key structural information loss. Here, we introduced the t-distributed stochastic neighbor embedding (t-SNE) method as a dimensionality reduction method with minimum structural information loss widely used in bioinformatics for analyses of macromolecules, especially biomacromolecules simulations. It is demonstrated that both one-dimensional (1D) and two-dimensional (2D) models of the t-SNE method are superior to distinguish important functional states of a model allosteric protein system for free energy and mechanistic analysis. Projections of the model protein simulations onto 1D and 2D t-SNE surfaces provide both clear visual cues and quantitative information, which is not readily available using other methods, regarding the transition mechanism between two important functional states of this protein.
中文翻译:

具有最小信息损失的大分子模拟t分布随机邻域嵌入方法
降维方法通常应用于大分子的分子动力学模拟,用于分析和可视化目的。通常希望合适的降维方法能够清楚地区分感兴趣系统具有不同构象的功能重要状态。然而,用于大分子模拟的常见降维方法,包括预定义的序参数和集体变量(CV)、主成分分析(PCA)和基于时间结构的独立成分分析(t-ICA),由于关键因素的影响而取得的成功有限。结构信息丢失。在这里,我们介绍了t分布随机邻域嵌入(t-SNE)方法作为一种具有最小结构信息损失的降维方法,广泛应用于生物信息学中的大分子分析,特别是生物大分子模拟。结果表明,t-SNE 方法的一维 (1D) 和二维 (2D) 模型均优于区分模型变构蛋白系统的重要功能状态,以进行自由能和机械分析。模型蛋白质模拟在一维和二维 t-SNE 表面上的投影提供了清晰的视觉线索和定量信息,这是使用其他方法不易获得的关于该蛋白质的两个重要功能状态之间的转换机制的信息。
更新日期:2018-09-25
中文翻译:
具有最小信息损失的大分子模拟t分布随机邻域嵌入方法
降维方法通常应用于大分子的分子动力学模拟,用于分析和可视化目的。通常希望合适的降维方法能够清楚地区分感兴趣系统具有不同构象的功能重要状态。然而,用于大分子模拟的常见降维方法,包括预定义的序参数和集体变量(CV)、主成分分析(PCA)和基于时间结构的独立成分分析(t-ICA),由于关键因素的影响而取得的成功有限。结构信息丢失。在这里,我们介绍了t分布随机邻域嵌入(t-SNE)方法作为一种具有最小结构信息损失的降维方法,广泛应用于生物信息学中的大分子分析,特别是生物大分子模拟。结果表明,t-SNE 方法的一维 (1D) 和二维 (2D) 模型均优于区分模型变构蛋白系统的重要功能状态,以进行自由能和机械分析。模型蛋白质模拟在一维和二维 t-SNE 表面上的投影提供了清晰的视觉线索和定量信息,这是使用其他方法不易获得的关于该蛋白质的两个重要功能状态之间的转换机制的信息。










































 京公网安备 11010802027423号
京公网安备 11010802027423号