当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural and Compositional Factors That Control the Li-Ion Conductivity in LiPON Electrolytes
Chemistry of Materials ( IF 7.2 ) Pub Date : 2018-09-15 00:00:00 , DOI: 10.1021/acs.chemmater.8b02812 Valentina Lacivita 1 , Nongnuch Artrith 2 , Gerbrand Ceder 1, 2
Chemistry of Materials ( IF 7.2 ) Pub Date : 2018-09-15 00:00:00 , DOI: 10.1021/acs.chemmater.8b02812 Valentina Lacivita 1 , Nongnuch Artrith 2 , Gerbrand Ceder 1, 2
Affiliation
|
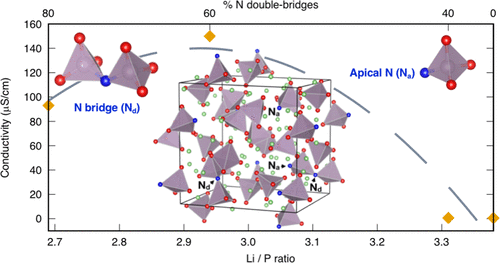
Amorphous Li-ion conductors are important solid-state electrolytes. However, Li transport in these systems is much less understood than for crystalline materials. We investigate amorphous LiPON electrolytes via ab initio molecular dynamics, providing atomistic-level insight into the mechanisms underlying the Li+ mobility. We find that the latter is strongly influenced by the chemistry and connectivity of phosphate polyanions near Li+. Amorphization generates edge-sharing polyhedral connections between Li(O,N)4 and P(O,N)4, and creates under- and overcoordinated Li sites, which destabilizes the Li+ and enhances their mobility. N substitution for O favors conductivity in two ways: (1) excess Li accompanying 1(N):1(O) substitutions introduces extra carriers; (2) energetically favored N-bridging substitutions condense phosphate units and densify the structure, which, counterintuitively, corresponds to higher Li+ mobility. Finally, bridging N is not only less electronegative than O but also engaged in strong covalent bonds with P. This weakens interactions with neighboring Li+ smoothing the way for their migration. When condensation of PO4 polyhedra leads to the formation of isolated O anions, the Li+ mobility is reduced, highlighting the importance of oxygen partial pressure control during synthesis. This detailed understanding of the structural mechanisms affecting Li+ mobility is the key for optimizing the conductivity of LiPON and other amorphous Li-ion conductors.
中文翻译:

控制LiPON电解质中锂离子电导率的结构和组成因素
非晶态锂离子导体是重要的固态电解质。但是,与结晶材料相比,人们对这些系统中的Li传输知之甚少。我们通过从头算分子动力学研究无定形LiPON电解质,提供有关Li +迁移率潜在机制的原子级见解。我们发现后者受到Li +附近磷酸根聚阴离子的化学性质和连通性的强烈影响。非晶化会在Li(O,N)4和P(O,N)4之间生成边缘共享的多面体连接,并会形成Li位过低和过高的Li位,这会破坏Li +的稳定性并增强他们的机动性。O的N取代以两种方式促进电导率:(1)伴随1(N):1(O)取代的过量Li引入了额外的载流子;(2)大力支持的N桥取代可缩合磷酸酯单元并致密化结构,这与直觉相反,对应于较高的Li +迁移率。最后,桥接N不仅比O具有负电性,而且还与P形成强共价键。这削弱了与相邻Li +的相互作用,为它们的迁移铺平了道路。当PO 4多面体的缩合导致形成孤立的O阴离子时,Li +迁移率降低,突出了合成过程中控制氧分压的重要性。对影响Li +迁移率的结构机理的详细了解是优化LiPON和其他非晶态锂离子导体导电性的关键。
更新日期:2018-09-15
中文翻译:
控制LiPON电解质中锂离子电导率的结构和组成因素
非晶态锂离子导体是重要的固态电解质。但是,与结晶材料相比,人们对这些系统中的Li传输知之甚少。我们通过从头算分子动力学研究无定形LiPON电解质,提供有关Li +迁移率潜在机制的原子级见解。我们发现后者受到Li +附近磷酸根聚阴离子的化学性质和连通性的强烈影响。非晶化会在Li(O,N)4和P(O,N)4之间生成边缘共享的多面体连接,并会形成Li位过低和过高的Li位,这会破坏Li +的稳定性并增强他们的机动性。O的N取代以两种方式促进电导率:(1)伴随1(N):1(O)取代的过量Li引入了额外的载流子;(2)大力支持的N桥取代可缩合磷酸酯单元并致密化结构,这与直觉相反,对应于较高的Li +迁移率。最后,桥接N不仅比O具有负电性,而且还与P形成强共价键。这削弱了与相邻Li +的相互作用,为它们的迁移铺平了道路。当PO 4多面体的缩合导致形成孤立的O阴离子时,Li +迁移率降低,突出了合成过程中控制氧分压的重要性。对影响Li +迁移率的结构机理的详细了解是优化LiPON和其他非晶态锂离子导体导电性的关键。










































 京公网安备 11010802027423号
京公网安备 11010802027423号