Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2018-09-14 , DOI: 10.1016/j.bioorg.2018.09.017 Ashraf A. Aly , Essmat M. El-Sheref , Momtaz E.M. Bakheet , Mai A.E. Mourad , Alan B. Brown , Stefan Bräse , Martin Nieger , Mahmoud A.A. Ibrahim
|
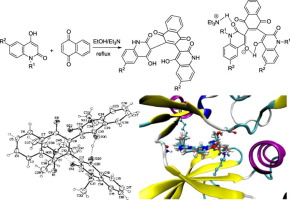
Two novel series of N-2,3-bis(6-substituted-4-hydroxy-2-oxo-1,2-dihydroquinolin-3-yl)naphthalene-1,4-diones 3a-d and substituted N-(methyl/ethyl)bisquinolinone triethyl-ammonium salts 4e,f were successfully synthesized. The synthesized compounds were targeted as new candidates to extracellular signal-regulated kinases 1/2 (ERK1/2) with considerable antineoplastic activity. The synthesis involved the reactions of 2 equivalents of 4-hydroxy-2(1H)-quinolinones 1a-f and one equivalent of 1,4-naphthoquinone (2) in a mixture of ethanol/dimethylformamide (1:1) as a solvent and 0.5 mL Et3N. In the reaction of 6-methyl-4-hydroxyquinolone 1b with 2, a side product 4b of the second series was obtained. In general, the presence of free NH-quinolone gave a single compound of the first series, whereas reaction of N-methyl/ethyl-quinolones 1e,f with 2 enhanced the formation of compounds of the second series. The structures of the new compounds were proved by different spectroscopic techniques such as IR, NMR (2D-NMR) and mass spectra, elemental analysis, and X-ray crystallography. To further elucidate the mechanism of action of these newly synthesized compounds, compounds 3a, 3b, 4e and 4f were selected to investigate for their MAP Kinases pathway inhibition together with molecular docking using ATP-binding site of ERK2. The results revealed that compounds 3a, 3b and 4f inhibited ETS-1 phosphorylation by ERK2 in a dose dependent manner. Also, compound 4f showed highest potency for ERK2 inhibition with ATP-competitive inhibition mechanism which was confirmed by the formation of three hydrogen bond in the molecular docking studies. The synthesized compounds were then tested for their in vitro anticancer activity against the NCI-60 panel of tumor cell lines. Interestingly, the selected compounds displayed from modest to strong cytotoxic activities. Compound 3b demonstrated broad spectrum anti-tumor activity against the nine tumor sub-panels tested, while compound 3d proved to be lethal to most of the cancer cell lines as shown by their promising GI50 and TGI values in NCI in vitro five dose testing. These results revealed that the synthesized compounds can potentially serve as leads for the development of novel chemotherapeutic agents and structure improvement will be necessary for some derivatives for enhancing their cellular activities and pharmacokinetic profile.
中文翻译:
新型1,2-双喹啉-1,4-萘醌的合成:ERK2抑制,细胞毒性和分子对接研究
N -2,3-双(6-取代的-4-羟基-2-羟基-2-氧代-1,2-二氢喹啉-3-基)萘-1,4-二酮3a-d和取代的N-(甲基)的两个新系列(乙基)双喹啉酮三乙基铵盐4e,f已成功合成。合成的化合物可作为具有明显抗肿瘤活性的细胞外信号调节激酶1/2(ERK1 / 2)的新候选物。合成涉及乙醇/二甲基甲酰胺(1:1)的混合物中的2当量的4-羟基-2(1 H)-喹啉酮1a - f和1当量的1,4-萘醌(2)的反应和0.5 mL Et 3N.在6-甲基-4-羟基喹诺酮1b与2的反应中,获得第二系列的副产物4b。通常,游离NH-喹诺酮的存在给出了第一系列的单一化合物,而N-甲基/乙基喹诺酮1e,f与2的反应增强了第二系列化合物的形成。通过不同的光谱技术,例如IR,NMR(2D-NMR)和质谱,元素分析和X射线晶体学,证明了新化合物的结构。为了进一步阐明这些新合成的化合物,化合物3a,3b,4e和4f的作用机理选择它们来研究其MAP激酶途径抑制以及使用ERK2 ATP结合位点的分子对接。结果表明,化合物3a,3b和4f以剂量依赖性方式抑制ERK2引起的ETS-1磷酸化。同样,化合物4f对ERK2的抑制作用最高,具有ATP竞争抑制机制,这在分子对接研究中通过形成三个氢键得到了证实。然后测试合成的化合物对肿瘤细胞系NCI-60的体外抗癌活性。有趣的是,所选化合物表现出从中等到强烈的细胞毒活性。化合物3b在体外五剂量试验中,化合物3d对NCI的潜在GI 50和TGI值显示出良好的抗GI活性,对9种被测试的肿瘤亚群具有广谱的抗肿瘤活性,而化合物3d被证明对大多数癌细胞具有致死性。这些结果表明,合成的化合物可以潜在地用作开发新型化学治疗剂的先导,并且对于某些衍生物以增强其细胞活性和药代动力学特性而言,结构改善将是必需的。


























 京公网安备 11010802027423号
京公网安备 11010802027423号