当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Multiscale Simulation Approach to Modeling Drug–Protein Binding Kinetics
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-13 00:00:00 , DOI: 10.1021/acs.jctc.8b00687 Susanta Haldar 1 , Federico Comitani , Giorgio Saladino , Christopher Woods 1 , Marc W van der Kamp 1, 2 , Adrian J Mulholland 1 , Francesco Luigi Gervasio
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-13 00:00:00 , DOI: 10.1021/acs.jctc.8b00687 Susanta Haldar 1 , Federico Comitani , Giorgio Saladino , Christopher Woods 1 , Marc W van der Kamp 1, 2 , Adrian J Mulholland 1 , Francesco Luigi Gervasio
Affiliation
|
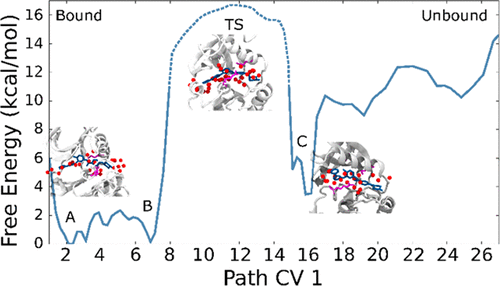
Drug–target binding kinetics has recently emerged as a sometimes critical determinant of in vivo efficacy and toxicity. Its rational optimization to improve potency or reduce side effects of drugs is, however, extremely difficult. Molecular simulations can play a crucial role in identifying features and properties of small ligands and their protein targets affecting the binding kinetics, but significant challenges include the long time scales involved in (un)binding events and the limited accuracy of empirical atomistic force fields (lacking, e.g., changes in electronic polarization). In an effort to overcome these hurdles, we propose a method that combines state-of-the-art enhanced sampling simulations and quantum mechanics/molecular mechanics (QM/MM) calculations at the BLYP/VDZ level to compute association free energy profiles and characterize the binding kinetics in terms of structure and dynamics of the transition state ensemble. We test our combined approach on the binding of the anticancer drug Imatinib to Src kinase, a well-characterized target for cancer therapy with a complex binding mechanism involving significant conformational changes. The results indicate significant changes in polarization along the binding pathways, which affect the predicted binding kinetics. This is likely to be of widespread importance in binding of ligands to protein targets.
中文翻译:

一种模拟药物-蛋白质结合动力学的多尺度模拟方法
药物-靶标结合动力学最近已成为体内有时关键的决定因素疗效和毒性。然而,对其进行合理优化以提高药效或减少药物副作用是极其困难的。分子模拟可以在识别小配体及其影响结合动力学的蛋白质靶点的特征和特性方面发挥关键作用,但重大挑战包括(非)结合事件涉及的长时间尺度和经验原子力场的有限准确性(缺乏,例如,电子极化的变化)。为了克服这些障碍,我们提出了一种方法,该方法结合了 BLYP/VDZ 级别的最先进的增强采样模拟和量子力学/分子力学 (QM/MM) 计算,以计算缔合自由能分布并表征过渡态系综的结构和动力学方面的结合动力学。我们测试了我们关于抗癌药物伊马替尼与 Src 激酶结合的组合方法,Src 激酶是一种具有良好特征的癌症治疗靶标,具有涉及显着构象变化的复杂结合机制。结果表明沿结合途径的极化发生显着变化,这会影响预测的结合动力学。这可能在配体与蛋白质靶标的结合中具有广泛的重要性。
更新日期:2018-09-13
中文翻译:
一种模拟药物-蛋白质结合动力学的多尺度模拟方法
药物-靶标结合动力学最近已成为体内有时关键的决定因素疗效和毒性。然而,对其进行合理优化以提高药效或减少药物副作用是极其困难的。分子模拟可以在识别小配体及其影响结合动力学的蛋白质靶点的特征和特性方面发挥关键作用,但重大挑战包括(非)结合事件涉及的长时间尺度和经验原子力场的有限准确性(缺乏,例如,电子极化的变化)。为了克服这些障碍,我们提出了一种方法,该方法结合了 BLYP/VDZ 级别的最先进的增强采样模拟和量子力学/分子力学 (QM/MM) 计算,以计算缔合自由能分布并表征过渡态系综的结构和动力学方面的结合动力学。我们测试了我们关于抗癌药物伊马替尼与 Src 激酶结合的组合方法,Src 激酶是一种具有良好特征的癌症治疗靶标,具有涉及显着构象变化的复杂结合机制。结果表明沿结合途径的极化发生显着变化,这会影响预测的结合动力学。这可能在配体与蛋白质靶标的结合中具有广泛的重要性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号