当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Exploratory machine-learned theoretical chemical shifts can closely predict metabolic mixture signals†
Chemical Science ( IF 7.6 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1039/c8sc03628d Kengo Ito 1, 2 , Yuka Obuchi 2 , Eisuke Chikayama 1, 3 , Yasuhiro Date 1, 2 , Jun Kikuchi 1, 2, 4
Chemical Science ( IF 7.6 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1039/c8sc03628d Kengo Ito 1, 2 , Yuka Obuchi 2 , Eisuke Chikayama 1, 3 , Yasuhiro Date 1, 2 , Jun Kikuchi 1, 2, 4
Affiliation
|
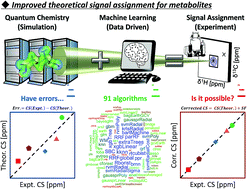
Various chemical shift predictive methodologies have been studied and developed, but there remains the problem of prediction accuracy. Assigning the NMR signals of metabolic mixtures requires high predictive performance owing to the complexity of the signals. Here we propose a new predictive tool that combines quantum chemistry and machine learning. A scaling factor as the objective variable to correct the errors of 2355 theoretical chemical shifts was optimized by exploring 91 machine learning algorithms and using the partial structure of 150 compounds as explanatory variables. The optimal predictive model gave RMSDs between experimental and predicted chemical shifts of 0.2177 ppm for δ1H and 3.3261 ppm for δ13C in the test data; thus, better accuracy was achieved compared with existing empirical and quantum chemical methods. The utility of the predictive model was demonstrated by applying it to assignments of experimental NMR signals of a complex metabolic mixture.
中文翻译:

探索性机器学习理论化学位移可以密切预测代谢混合物信号†
人们已经研究和开发了各种化学位移预测方法,但仍然存在预测准确性的问题。由于信号的复杂性,分配代谢混合物的 NMR 信号需要高预测性能。在这里,我们提出了一种结合量子化学和机器学习的新预测工具。通过探索91种机器学习算法并使用150种化合物的部分结构作为解释变量,优化了作为目标变量的比例因子来校正2355个理论化学位移的误差。最佳预测模型给出了测试数据中δ 1 H 的实验化学位移和预测化学位移之间的 RMSD 为 0.2177 ppm 和δ 13 C 的 3.3261 ppm;因此,与现有的经验和量子化学方法相比,获得了更好的准确性。通过将预测模型应用于复杂代谢混合物的实验 NMR 信号分配,证明了该预测模型的实用性。
更新日期:2018-09-10
中文翻译:
探索性机器学习理论化学位移可以密切预测代谢混合物信号†
人们已经研究和开发了各种化学位移预测方法,但仍然存在预测准确性的问题。由于信号的复杂性,分配代谢混合物的 NMR 信号需要高预测性能。在这里,我们提出了一种结合量子化学和机器学习的新预测工具。通过探索91种机器学习算法并使用150种化合物的部分结构作为解释变量,优化了作为目标变量的比例因子来校正2355个理论化学位移的误差。最佳预测模型给出了测试数据中δ 1 H 的实验化学位移和预测化学位移之间的 RMSD 为 0.2177 ppm 和δ 13 C 的 3.3261 ppm;因此,与现有的经验和量子化学方法相比,获得了更好的准确性。通过将预测模型应用于复杂代谢混合物的实验 NMR 信号分配,证明了该预测模型的实用性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号