当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Simple Correction for Nonadditive Dispersion within Extended Symmetry-Adapted Perturbation Theory (XSAPT)
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1021/acs.jctc.8b00527 Ka Un Lao 1 , John M. Herbert 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1021/acs.jctc.8b00527 Ka Un Lao 1 , John M. Herbert 1
Affiliation
|
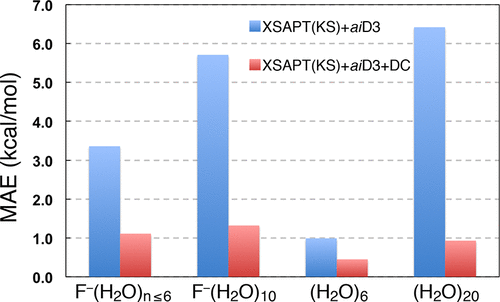
Extended symmetry-adapted perturbation theory (XSAPT), in conjunction with empirical “+aiD” potentials fit to ab initio dispersion data, is a low-scaling approach to compute intermolecular interaction energies in noncovalent clusters. One shortcoming is that the aiD atom–atom dispersion potentials are independent of the chemical environment of the atoms in question and therefore neglect nonadditive dispersion effects. These can be significant in large systems, so to account for them we test a simple correction to XSAPT(KS)+aiD, where “KS” indicates the use of Kohn–Sham orbitals. This correction, which can be evaluated at fourth-order cost using double-ζ basis sets, is based on comparing second-order SAPT dispersion with and without a self-consistent charge embedding for the monomer wave functions. The correction amounts to ∼1.4 kcal/mol in (H2O)6 but ∼5.5 kcal/mol in (H2O)20. With the nonadditive dispersion correction, XSAPT(KS)+aiD affords errors of ∼1 kcal/mol for isomers of F–(H2O)10 and (H2O)20, where the benchmarks are complete-basis CCSD(T) energies, as well as for ion–water clusters X(H2O)n where n ≤ 6 and X = F–, Cl–, SO42–, Li+, Na+, or K+. We also test the MP2 method and a variety of density-functional methods that have been specifically recommended for noncovalent interactions. Among the latter, only ωB97X-V and ωB97M-V can be recommended for ion–water clusters, as mean errors for other popular approaches (including ωB97X-D3 and several Minnesota functionals) exceed 1 kcal/mol. Lastly, we examine clathrate-hydrate host/guest complexes whose mixture of hydrogen bonding and dispersion make them challenging tests for noncovalent quantum chemistry. Although the B97-D2 functional performs best for clathrate hydrates and has been previously recommended in other studies of these inclusion complexes, its performance for other systems examined here is quite poor. We are unable to find a functional whose accuracy is ≲1 kcal/mol accuracy for both clathrate hydrates and ion–water clusters. However, the XSAPT(KS)+aiD method with the nonadditive dispersion correction can achieve this, with a mean error for the clathrate hydrates of 0.3 kcal/mol.
中文翻译:

扩展对称适应扰动理论(XSAPT)中非加性色散的简单校正
扩展的对称自适应扰动理论(XSAPT)与经验“ + ai D”电势相符,适合从头算起的散布数据,是一种计算非共价簇中分子间相互作用能的低尺度方法。一个缺点是,ai D原子-原子的弥散势与所讨论原子的化学环境无关,因此忽略了非加性弥散效应。这些在大型系统中可能很重要,因此,为解决这些问题,我们测试了对XSAPT(KS)+ ai的简单修正D,其中“ KS”表示使用Kohn–Sham轨道。可以使用双ζ基集以四阶成本评估此校正,它是基于比较带有和不带有针对单体波函数的自洽电荷嵌入的二阶SAPT色散。校正量在(H 2 O)6中约为1.4 kcal / mol,而在(H 2 O)20中约为5.5 kcal / mol 。通过非相加色散校正,XSAPT(KS)+ ai D对于F –(H 2 O)10和(H 2 O)20的异构体提供的误差约为1 kcal / mol ,其中基准是完全基准的CCSD(T )能量以及离子水簇X(H2 O)ñ其中Ñ ≤6和X = F - ,氯-,SO 4 2-,李+,钠+,或K +。我们还测试了MP2方法和特别推荐用于非共价相互作用的各种密度泛函方法。在后者中,仅推荐将ωB97X-V和ωB97M-V用于离子水团簇,因为其他流行方法(包括ωB97X-D3和一些明尼苏达州官能团)的平均误差超过1 kcal / mol。最后,我们研究了笼形水合物主体/客体复合物,其氢键和分散体的混合使其成为非共价量子化学的具有挑战性的测试。尽管B97-D2官能团对于笼形水合物表现最佳,并且先前在对这些包合物的其他研究中已被推荐使用,但对于此处研究的其他系统,其性能却很差。对于包合物水合物和离子水簇,我们找不到精确度accuracy1 kcal / mol的功能。采用非累加色散校正的ai D方法可以实现这一点,而笼形水合物的平均误差为0.3 kcal / mol。
更新日期:2018-09-10
中文翻译:
扩展对称适应扰动理论(XSAPT)中非加性色散的简单校正
扩展的对称自适应扰动理论(XSAPT)与经验“ + ai D”电势相符,适合从头算起的散布数据,是一种计算非共价簇中分子间相互作用能的低尺度方法。一个缺点是,ai D原子-原子的弥散势与所讨论原子的化学环境无关,因此忽略了非加性弥散效应。这些在大型系统中可能很重要,因此,为解决这些问题,我们测试了对XSAPT(KS)+ ai的简单修正D,其中“ KS”表示使用Kohn–Sham轨道。可以使用双ζ基集以四阶成本评估此校正,它是基于比较带有和不带有针对单体波函数的自洽电荷嵌入的二阶SAPT色散。校正量在(H 2 O)6中约为1.4 kcal / mol,而在(H 2 O)20中约为5.5 kcal / mol 。通过非相加色散校正,XSAPT(KS)+ ai D对于F –(H 2 O)10和(H 2 O)20的异构体提供的误差约为1 kcal / mol ,其中基准是完全基准的CCSD(T )能量以及离子水簇X(H2 O)ñ其中Ñ ≤6和X = F - ,氯-,SO 4 2-,李+,钠+,或K +。我们还测试了MP2方法和特别推荐用于非共价相互作用的各种密度泛函方法。在后者中,仅推荐将ωB97X-V和ωB97M-V用于离子水团簇,因为其他流行方法(包括ωB97X-D3和一些明尼苏达州官能团)的平均误差超过1 kcal / mol。最后,我们研究了笼形水合物主体/客体复合物,其氢键和分散体的混合使其成为非共价量子化学的具有挑战性的测试。尽管B97-D2官能团对于笼形水合物表现最佳,并且先前在对这些包合物的其他研究中已被推荐使用,但对于此处研究的其他系统,其性能却很差。对于包合物水合物和离子水簇,我们找不到精确度accuracy1 kcal / mol的功能。采用非累加色散校正的ai D方法可以实现这一点,而笼形水合物的平均误差为0.3 kcal / mol。










































 京公网安备 11010802027423号
京公网安备 11010802027423号