当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
QM/MM Calculations on Protein–RNA Complexes: Understanding Limitations of Classical MD Simulations and Search for Reliable Cost-Effective QM Methods
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1021/acs.jctc.8b00670 Pavlína Pokorná 1 , Holger Kruse 1 , Miroslav Krepl 1 , Jiří Šponer 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-10 00:00:00 , DOI: 10.1021/acs.jctc.8b00670 Pavlína Pokorná 1 , Holger Kruse 1 , Miroslav Krepl 1 , Jiří Šponer 1, 2
Affiliation
|
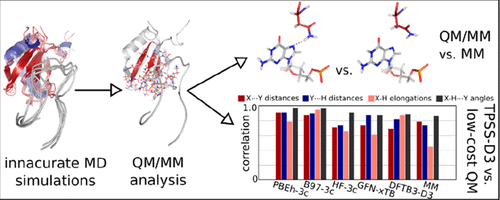
Although atomistic explicit-solvent Molecular Dynamics (MD) is a popular tool to study protein–RNA recognition, satisfactory MD description of protein–RNA complexes is not always achieved. Unfortunately, it is often difficult to separate MD simulation instabilities primarily caused by the simple point-charge molecular mechanics (MM) force fields from problems related to the notorious uncertainties in the starting structures. Herein, we report a series of large-scale QM/MM calculations on the U1A protein–RNA complex. This experimentally well-characterized system has an intricate protein–RNA interface, which is very unstable in MD simulations. The QM/MM calculations identify several H-bonds poorly described by the MM method and thus indicate the sources of instabilities of the U1A interface in MD simulations. The results suggest that advanced QM/MM computations could be used to indirectly rationalize problems seen in MM-based MD simulations of protein–RNA complexes. As the most accurate QM method, we employ the computationally demanding meta-GGA density functional TPSS-D3(BJ)/def2-TZVP level of theory. Because considerably faster methods would be needed to extend sampling and to study even larger protein–RNA interfaces, a set of low-cost QM/MM methods is compared to the TPSS-D3(BJ)/def2-TZVP data. The PBEh-3c and B97-3c density functional composite methods appear to be suitable for protein–RNA interfaces. In contrast, HF-3c and the tight-binding Hamiltonians DFTB3-D3 and GFN-xTB perform unsatisfactorily and do not provide any advantage over the MM description. These conclusions are supported also by similar analysis of a simple HutP protein–RNA interface, which is well-described by MD with the exception of just one H-bond. Some other methodological aspects of QM/MM calculations on protein–RNA interfaces are discussed.
中文翻译:

蛋白质-RNA复合物的QM / MM计算:了解经典MD模拟的局限性并寻找可靠的具有成本效益的QM方法
尽管原子显式溶剂分子动力学(MD)是研究蛋白质-RNA识别的一种流行工具,但是蛋白质-RNA复合物的MD描述不一定总是令人满意的。不幸的是,通常很难将主要由简单的点电荷分子力学(MM)力场引起的MD模拟不稳定性与与起始结构中众所周知的不确定性相关的问题区分开来。在此,我们报告了有关U1A蛋白质-RNA复合物的一系列大规模QM / MM计算。这个实验特性良好的系统具有复杂的蛋白质-RNA界面,在MD模拟中非常不稳定。QM / MM计算可识别出几种用MM方法描述不佳的H键,从而指出了MD模拟中U1A接口不稳定性的根源。结果表明,先进的QM / MM计算可用于间接合理化蛋白质-RNA复合物基于MM的MD模拟中看到的问题。作为最准确的质量管理方法,我们采用了计算要求严格的meta-GGA密度函数TPSS-D3(BJ)/ def2-TZVP的理论水平。由于将需要相当快的方法来扩展采样并研究更大的蛋白质-RNA界面,因此将一组低成本QM / MM方法与TPSS-D3(BJ)/ def2-TZVP数据进行了比较。PBEh-3c和B97-3c密度泛函复合方法似乎适用于蛋白质-RNA界面。相反,HF-3c和紧密结合的哈密顿量DFTB3-D3和GFN-xTB的性能不能令人满意,与MM描述相比没有任何优势。这些结论也得到了对简单的HutP蛋白质-RNA界面的类似分析的支持,该现象由MD很好地描述,只有一个H键除外。讨论了蛋白质-RNA界面上QM / MM计算的其他方法论方面。
更新日期:2018-09-10
中文翻译:
蛋白质-RNA复合物的QM / MM计算:了解经典MD模拟的局限性并寻找可靠的具有成本效益的QM方法
尽管原子显式溶剂分子动力学(MD)是研究蛋白质-RNA识别的一种流行工具,但是蛋白质-RNA复合物的MD描述不一定总是令人满意的。不幸的是,通常很难将主要由简单的点电荷分子力学(MM)力场引起的MD模拟不稳定性与与起始结构中众所周知的不确定性相关的问题区分开来。在此,我们报告了有关U1A蛋白质-RNA复合物的一系列大规模QM / MM计算。这个实验特性良好的系统具有复杂的蛋白质-RNA界面,在MD模拟中非常不稳定。QM / MM计算可识别出几种用MM方法描述不佳的H键,从而指出了MD模拟中U1A接口不稳定性的根源。结果表明,先进的QM / MM计算可用于间接合理化蛋白质-RNA复合物基于MM的MD模拟中看到的问题。作为最准确的质量管理方法,我们采用了计算要求严格的meta-GGA密度函数TPSS-D3(BJ)/ def2-TZVP的理论水平。由于将需要相当快的方法来扩展采样并研究更大的蛋白质-RNA界面,因此将一组低成本QM / MM方法与TPSS-D3(BJ)/ def2-TZVP数据进行了比较。PBEh-3c和B97-3c密度泛函复合方法似乎适用于蛋白质-RNA界面。相反,HF-3c和紧密结合的哈密顿量DFTB3-D3和GFN-xTB的性能不能令人满意,与MM描述相比没有任何优势。这些结论也得到了对简单的HutP蛋白质-RNA界面的类似分析的支持,该现象由MD很好地描述,只有一个H键除外。讨论了蛋白质-RNA界面上QM / MM计算的其他方法论方面。










































 京公网安备 11010802027423号
京公网安备 11010802027423号