Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experimental and computational framework for a dynamic protein atlas of human cell division
Nature ( IF 64.8 ) Pub Date : 2018-09-01 , DOI: 10.1038/s41586-018-0518-z Yin Cai , M. Julius Hossain , Jean-Karim Hériché , Antonio Z. Politi , Nike Walther , Birgit Koch , Malte Wachsmuth , Bianca Nijmeijer , Moritz Kueblbeck , Marina Martinic-Kavur , Rene Ladurner , Stephanie Alexander , Jan-Michael Peters , Jan Ellenberg
Nature ( IF 64.8 ) Pub Date : 2018-09-01 , DOI: 10.1038/s41586-018-0518-z Yin Cai , M. Julius Hossain , Jean-Karim Hériché , Antonio Z. Politi , Nike Walther , Birgit Koch , Malte Wachsmuth , Bianca Nijmeijer , Moritz Kueblbeck , Marina Martinic-Kavur , Rene Ladurner , Stephanie Alexander , Jan-Michael Peters , Jan Ellenberg
|
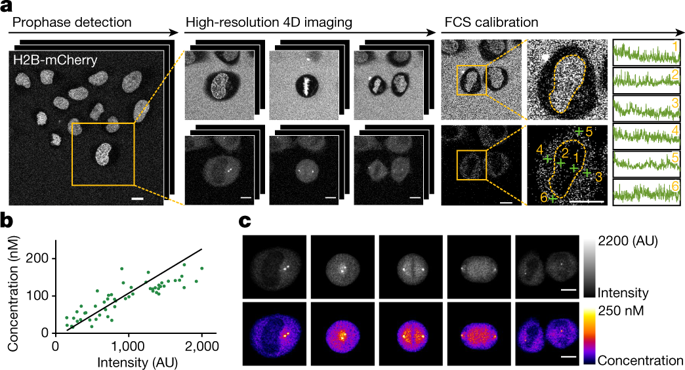
Essential biological functions, such as mitosis, require tight coordination of hundreds of proteins in space and time. Localization, the timing of interactions and changes in cellular structure are all crucial to ensure the correct assembly, function and regulation of protein complexes1–4. Imaging of live cells can reveal protein distributions and dynamics but experimental and theoretical challenges have prevented the collection of quantitative data, which are necessary for the formulation of a model of mitosis that comprehensively integrates information and enables the analysis of the dynamic interactions between the molecular parts of the mitotic machinery within changing cellular boundaries. Here we generate a canonical model of the morphological changes during the mitotic progression of human cells on the basis of four-dimensional image data. We use this model to integrate dynamic three-dimensional concentration data of many fluorescently knocked-in mitotic proteins, imaged by fluorescence correlation spectroscopy-calibrated microscopy5. The approach taken here to generate a dynamic protein atlas of human cell division is generic; it can be applied to systematically map and mine dynamic protein localization networks that drive cell division in different cell types, and can be conceptually transferred to other cellular functions.Quantitative live-cell imaging provides a dynamic protein atlas of mitosis.
中文翻译:

人类细胞分裂动态蛋白质图谱的实验和计算框架
基本的生物学功能,例如有丝分裂,需要数百种蛋白质在空间和时间上的紧密协调。定位、相互作用的时间和细胞结构的变化都是确保蛋白质复合物正确组装、功能和调节的关键1-4。活细胞成像可以揭示蛋白质分布和动力学,但实验和理论挑战阻碍了定量数据的收集,这对于制定全面整合信息并能够分析分子部分之间的动态相互作用的有丝分裂模型是必要的在不断变化的细胞边界内的有丝分裂机制。在这里,我们基于四维图像数据生成了人类细胞有丝分裂过程中形态学变化的典型模型。我们使用这个模型来整合许多荧光敲入有丝分裂蛋白的动态三维浓度数据,通过荧光相关光谱校准显微镜成像5。这里用来生成人类细胞分裂动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。这里用来生成人类细胞分裂的动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。这里用来生成人类细胞分裂动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。
更新日期:2018-09-01
中文翻译:
人类细胞分裂动态蛋白质图谱的实验和计算框架
基本的生物学功能,例如有丝分裂,需要数百种蛋白质在空间和时间上的紧密协调。定位、相互作用的时间和细胞结构的变化都是确保蛋白质复合物正确组装、功能和调节的关键1-4。活细胞成像可以揭示蛋白质分布和动力学,但实验和理论挑战阻碍了定量数据的收集,这对于制定全面整合信息并能够分析分子部分之间的动态相互作用的有丝分裂模型是必要的在不断变化的细胞边界内的有丝分裂机制。在这里,我们基于四维图像数据生成了人类细胞有丝分裂过程中形态学变化的典型模型。我们使用这个模型来整合许多荧光敲入有丝分裂蛋白的动态三维浓度数据,通过荧光相关光谱校准显微镜成像5。这里用来生成人类细胞分裂动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。这里用来生成人类细胞分裂的动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。这里用来生成人类细胞分裂动态蛋白质图谱的方法是通用的。它可以用于系统地绘制和挖掘在不同细胞类型中驱动细胞分裂的动态蛋白质定位网络,并且可以在概念上转移到其他细胞功能。定量活细胞成像提供了有丝分裂的动态蛋白质图谱。


























 京公网安备 11010802027423号
京公网安备 11010802027423号