当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electronic Energies Are Not Enough: An Ion Mobility-Aided, Quantum Chemical Benchmark Analysis of H+GPGG Conformers
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-07 00:00:00 , DOI: 10.1021/acs.jctc.8b00648 Daniel Beckett 1 , Tarick J. El-Baba 1 , David E. Clemmer 1 , Krishnan Raghavachari 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-09-07 00:00:00 , DOI: 10.1021/acs.jctc.8b00648 Daniel Beckett 1 , Tarick J. El-Baba 1 , David E. Clemmer 1 , Krishnan Raghavachari 1
Affiliation
|
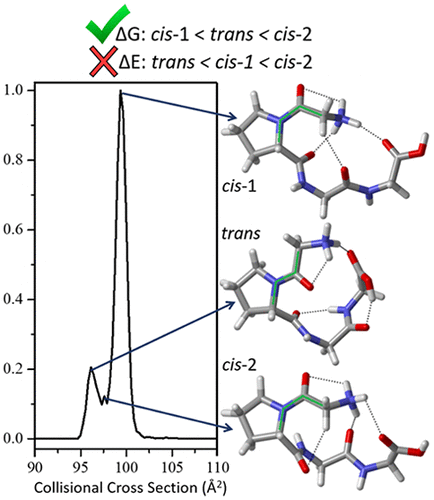
Peptides and proteins exist not as single, lowest energy structures but as ensembles of states separated by small barriers. In order to study these species we must be able to correctly identify their gas phase conformational distributions, and ion mobility spectrometry (IMS) has arisen as an experimental method for assessing the gas phase energetics of flexible peptides. Here, we present a thorough exploration and benchmarking of the low energy conformers of the small, hairpin peptide H+GPGG with the aid of ion mobility spectrometry against a wide swath of density functionals (35 dispersion-corrected and uncorrected functionals represented by 21 unique exchange–correlation functionals) and wave function theory methods (15 total levels of theory). The three experimentally resolved IMS peaks were found to correspond to three distinct pairs of conformers, each pair composed of species differing only by the chair or boat configuration of the proline. Two of the H+GPGG conformer pairs possess a cis configuration about the Pro–Gly1 peptide bond while the other adopts a trans configuration. While the experimental spectrum reports a higher intensity for the cis-1 conformer than the trans conformer, 13 WFT levels of theory, including a complete basis set CCSD(T) extrapolation, obtain trans to be favored in terms of the electronic energy. This same effect is seen in 14 of the 18 dispersion-corrected density functionals studied, whereas the remaining 17 functionals show more variety. Only when Gibbs free energies are considered do the WFT methods and dispersion-corrected functionals reflect the experimental distribution. CAM-B3LYP-D3BJ emerges as the best-performing density functional, matching the experimental distribution and the CCSD(T)/CBS relative energies within 6%. Further analysis reveals the trans conformer to be favored electronically, but entropically disfavored, leading to the experimental preference of the cis-1 conformer. These results highlight the danger in considering only the electronic energies, which is common practice in electronic structure theory predictions of conformational energy distributions. Additionally, the effects of temperature and scaling of the frequencies used in obtaining the Gibbs free energies are explored.
中文翻译:

电子能量不足:H + GPGG构象异构体的离子迁移辅助量子化学基准分析
肽和蛋白质并非以单一的,最低能量的结构存在,而是以小障碍隔开的状态的集合形式存在。为了研究这些物种,我们必须能够正确识别它们的气相构象分布,并且离子迁移谱法(IMS)已经成为评估柔性肽的气相能量的实验方法。在这里,我们对发夹小肽H +的低能构象体进行了全面的探索和基准测试GPGG借助离子迁移谱仪针对大量密度泛函(由21种独特的交换-相关泛函表示的35种弥散校正和未校正的泛函)和波动函数理论方法(总共15种理论水平)进行了分析。发现三个通过实验解析的IMS峰对应于三对不同的构象异构体,每对构象异构体仅因脯氨酸的椅子或船形而异。H + GPGG构象对中的两个具有Pro-Gly 1肽键的顺式构型,而另一个则具有反式构型。虽然实验光谱报告的顺式-1构象异构体强度比顺式构象的强度高。反式构象异构体,具有13种WFT理论水平,包括一个完整的基础CCSD(T)外推法,可以得到反式电子能量,因此受到青睐。在所研究的18种色散校正密度官能团中,有14种具有相同的效果,而其余17种官能团则表现出更多的多样性。仅当考虑吉布斯自由能时,WFT方法和色散校正的功能才可以反映实验分布。CAM-B3LYP-D3BJ成为表现最佳的密度泛函,与实验分布和CCSD(T)/ CBS相对能量在6%之内相匹配。进一步的分析表明反式构象异构体在电子上受到青睐,但在熵方面却不受欢迎,从而导致顺式的实验偏爱-1构象体。这些结果突出了仅考虑电子能量的危险,这是构象能量分布的电子结构理论预测中的常见实践。另外,探索了温度和获得吉布斯自由能所用频率的缩放比例的影响。
更新日期:2018-09-07
中文翻译:
电子能量不足:H + GPGG构象异构体的离子迁移辅助量子化学基准分析
肽和蛋白质并非以单一的,最低能量的结构存在,而是以小障碍隔开的状态的集合形式存在。为了研究这些物种,我们必须能够正确识别它们的气相构象分布,并且离子迁移谱法(IMS)已经成为评估柔性肽的气相能量的实验方法。在这里,我们对发夹小肽H +的低能构象体进行了全面的探索和基准测试GPGG借助离子迁移谱仪针对大量密度泛函(由21种独特的交换-相关泛函表示的35种弥散校正和未校正的泛函)和波动函数理论方法(总共15种理论水平)进行了分析。发现三个通过实验解析的IMS峰对应于三对不同的构象异构体,每对构象异构体仅因脯氨酸的椅子或船形而异。H + GPGG构象对中的两个具有Pro-Gly 1肽键的顺式构型,而另一个则具有反式构型。虽然实验光谱报告的顺式-1构象异构体强度比顺式构象的强度高。反式构象异构体,具有13种WFT理论水平,包括一个完整的基础CCSD(T)外推法,可以得到反式电子能量,因此受到青睐。在所研究的18种色散校正密度官能团中,有14种具有相同的效果,而其余17种官能团则表现出更多的多样性。仅当考虑吉布斯自由能时,WFT方法和色散校正的功能才可以反映实验分布。CAM-B3LYP-D3BJ成为表现最佳的密度泛函,与实验分布和CCSD(T)/ CBS相对能量在6%之内相匹配。进一步的分析表明反式构象异构体在电子上受到青睐,但在熵方面却不受欢迎,从而导致顺式的实验偏爱-1构象体。这些结果突出了仅考虑电子能量的危险,这是构象能量分布的电子结构理论预测中的常见实践。另外,探索了温度和获得吉布斯自由能所用频率的缩放比例的影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号