当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Coupled Cluster and Time-Dependent Density Functional Theory Description of Inner Shell Photoabsorption Cross Sections of Molecules
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-06 00:00:00 , DOI: 10.1021/acs.jctc.8b00375 Bruno Nunes Cabral Tenorio 1 , Ricardo Rodrigues Oliveira 1 , Marco Antonio Chaer Nascimento 1 , Alexandre Braga Rocha 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-09-06 00:00:00 , DOI: 10.1021/acs.jctc.8b00375 Bruno Nunes Cabral Tenorio 1 , Ricardo Rodrigues Oliveira 1 , Marco Antonio Chaer Nascimento 1 , Alexandre Braga Rocha 1
Affiliation
|
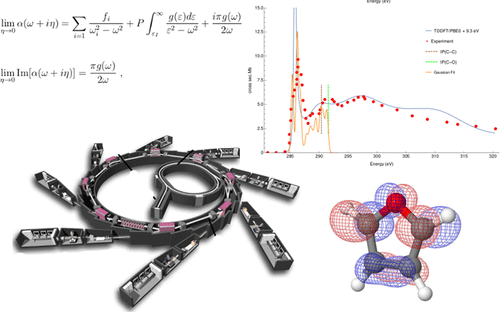
Near K-edge photoabsorption cross section spectra of a number of molecules, namely, water, ammonia, acetone, acetaldehyde, furan, and pyrrole, were obtained at the nitrogen, oxygen, and carbon K-edges with the Coupled Cluster ansatz (CC) and with the Time-Dependent Density Functional Theory (TDDFT) by treating the inner shell excitations as individual channels, separated from the valence part of the spectrum. The discretized electronic pseudospectrum, obtained with quadratically integrable basis sets (a.k.a. L2) at the CC or TDDFT level, is used to reconstruct the complex dipole polarizability function, from which the photoabsorption cross section near the K-edge is obtained by a continued fraction based analytic continuation procedure. The CC2 and CCSD results are in good agreement with experimental data, while the TDDFT results yield reliable cross sections. Overall, the results obtained in this work indicate that our method can be used for the treatment of the NEXAFS spectra, with the advantage that the electronic excitations in the K-edges can be easily obtained at low computational cost using TDDFT.
中文翻译:

分子内壳光吸收截面的簇和时变密度泛函理论耦合描述
利用耦合簇ansatz(CC)在氮,氧和碳K边缘获得了许多分子(即水,氨,丙酮,乙醛,呋喃和吡咯)的近K边缘光吸收截面光谱。并使用时变密度泛函理论(TDDFT)将内壳激发视为独立的通道,与光谱的价态部分分开。离散电子伪谱,通过二次可积基集(aka L 2)在CC或TDDFT级别用于重建复偶极子极化率函数,通过基于连续分数的解析连续过程从中获得K边缘附近的光吸收截面。CC2和CCSD结果与实验数据非常吻合,而TDDFT结果产生可靠的横截面。总的来说,这项工作中获得的结果表明我们的方法可以用于NEXAFS光谱的处理,其优点是可以使用TDDFT以较低的计算成本轻松获得K边缘中的电子激发。
更新日期:2018-09-06
中文翻译:
分子内壳光吸收截面的簇和时变密度泛函理论耦合描述
利用耦合簇ansatz(CC)在氮,氧和碳K边缘获得了许多分子(即水,氨,丙酮,乙醛,呋喃和吡咯)的近K边缘光吸收截面光谱。并使用时变密度泛函理论(TDDFT)将内壳激发视为独立的通道,与光谱的价态部分分开。离散电子伪谱,通过二次可积基集(aka L 2)在CC或TDDFT级别用于重建复偶极子极化率函数,通过基于连续分数的解析连续过程从中获得K边缘附近的光吸收截面。CC2和CCSD结果与实验数据非常吻合,而TDDFT结果产生可靠的横截面。总的来说,这项工作中获得的结果表明我们的方法可以用于NEXAFS光谱的处理,其优点是可以使用TDDFT以较低的计算成本轻松获得K边缘中的电子激发。


























 京公网安备 11010802027423号
京公网安备 11010802027423号