当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The surface and catalytic chemistry of the first row transition metal phosphides in deoxygenation†
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2018-09-06 00:00:00 , DOI: 10.1039/c8cy01134f Yang He 1, 2, 3, 4 , Siris Laursen 1, 2, 3, 4
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2018-09-06 00:00:00 , DOI: 10.1039/c8cy01134f Yang He 1, 2, 3, 4 , Siris Laursen 1, 2, 3, 4
Affiliation
|
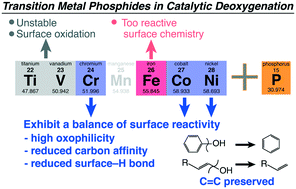
The proven utility of transition metal (TM) carbides, sulfides, and phosphides in catalytic deoxygenation reactions and their ability to preserve unsaturation or aromaticity in products has suggested the materials exhibit unique surface chemistry towards C, O, and H that is inaccessible when using reduced metal or TM + TM alloy catalysts. Herein, we present a computational surface chemistry study of the deoxygenation of phenol over most 1st row TM phosphides. Reaction mechanism analysis showed that dramatically enhanced surface reactivity towards oxygen was responsible for driving C–O cleavage. Reduced surface chemical reactivity towards carbon and limited hydrogenation activity were both beneficial in limiting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C activation in the aromatic ring and unselective overhydrogenation. The more covalent bonding within the phosphides inhibited a correlation between kinetics and thermodynamics of the C–O cleavage step due to the energetics associated with electron density transfer to or from the catalyst surface. Hydrogenation, a mostly covalent reaction step, tracked well with surface reactivity markers and d-band center of the phosphides. The less metallic electronic structure also contributed to electronically different hydrogen bonding to the surface and limited kinetics for hydrogenation that may favorably reduce unselective CC hydrogenation. Surface reaction site nature also tracked with bonding within the phosphides and their metal-to-nonmetal ratio suggesting strong electronic effects in the manipulation of the metal reaction sites and the role of nonmetal sites in the reaction mechanism.
C activation in the aromatic ring and unselective overhydrogenation. The more covalent bonding within the phosphides inhibited a correlation between kinetics and thermodynamics of the C–O cleavage step due to the energetics associated with electron density transfer to or from the catalyst surface. Hydrogenation, a mostly covalent reaction step, tracked well with surface reactivity markers and d-band center of the phosphides. The less metallic electronic structure also contributed to electronically different hydrogen bonding to the surface and limited kinetics for hydrogenation that may favorably reduce unselective CC hydrogenation. Surface reaction site nature also tracked with bonding within the phosphides and their metal-to-nonmetal ratio suggesting strong electronic effects in the manipulation of the metal reaction sites and the role of nonmetal sites in the reaction mechanism.
中文翻译:

脱氧中第一排过渡金属磷化物的表面和催化化学作用†
过渡金属(TM)碳化物,硫化物和磷化物在催化脱氧反应中的实用性及其在产品中保留不饱和或芳香性的能力表明,该材料对C,O和H表现出独特的表面化学性质,当使用金属或TM + TM合金催化剂。在这里,我们介绍了在大多数第一排TM磷化物上苯酚的脱氧的计算表面化学研究。反应机理分析表明,表面对氧气的反应性显着增强是推动C-O裂解的原因。降低对碳的表面化学反应性和有限的氢化活性均有利于限制C芳环中的C活化和非选择性过度氢化。由于与电子密度转移到催化剂表面或从催化剂表面转移出来的能量有关,磷化物中更共价的键抑制了C-O裂解步骤的动力学和热力学之间的相关性。氢化是一种大多数为共价反应的步骤,可通过表面反应性标记和磷的d带中心很好地跟踪。较少金属的电子结构还有助于在表面上以电子方式不同的氢键和有限的氢化动力学,可能有利地降低非选择性CC氢化。表面反应位点的性质也随磷化物内的键合及其金属与非金属比的变化而变化,这表明在操纵金属反应位点和非金属位点在反应机理中的作用方面具有很强的电子效应。
更新日期:2018-09-06
C activation in the aromatic ring and unselective overhydrogenation. The more covalent bonding within the phosphides inhibited a correlation between kinetics and thermodynamics of the C–O cleavage step due to the energetics associated with electron density transfer to or from the catalyst surface. Hydrogenation, a mostly covalent reaction step, tracked well with surface reactivity markers and d-band center of the phosphides. The less metallic electronic structure also contributed to electronically different hydrogen bonding to the surface and limited kinetics for hydrogenation that may favorably reduce unselective CC hydrogenation. Surface reaction site nature also tracked with bonding within the phosphides and their metal-to-nonmetal ratio suggesting strong electronic effects in the manipulation of the metal reaction sites and the role of nonmetal sites in the reaction mechanism.
中文翻译:
脱氧中第一排过渡金属磷化物的表面和催化化学作用†
过渡金属(TM)碳化物,硫化物和磷化物在催化脱氧反应中的实用性及其在产品中保留不饱和或芳香性的能力表明,该材料对C,O和H表现出独特的表面化学性质,当使用金属或TM + TM合金催化剂。在这里,我们介绍了在大多数第一排TM磷化物上苯酚的脱氧的计算表面化学研究。反应机理分析表明,表面对氧气的反应性显着增强是推动C-O裂解的原因。降低对碳的表面化学反应性和有限的氢化活性均有利于限制C
芳环中的C活化和非选择性过度氢化。由于与电子密度转移到催化剂表面或从催化剂表面转移出来的能量有关,磷化物中更共价的键抑制了C-O裂解步骤的动力学和热力学之间的相关性。氢化是一种大多数为共价反应的步骤,可通过表面反应性标记和磷的d带中心很好地跟踪。较少金属的电子结构还有助于在表面上以电子方式不同的氢键和有限的氢化动力学,可能有利地降低非选择性CC氢化。表面反应位点的性质也随磷化物内的键合及其金属与非金属比的变化而变化,这表明在操纵金属反应位点和非金属位点在反应机理中的作用方面具有很强的电子效应。










































 京公网安备 11010802027423号
京公网安备 11010802027423号