Journal of the American Society for Mass Spectrometry ( IF 3.1 ) Pub Date : 2018-08-31 , DOI: 10.1007/s13361-018-2056-1 Lars Sørensen 1, 2 , Rune Salbo 3
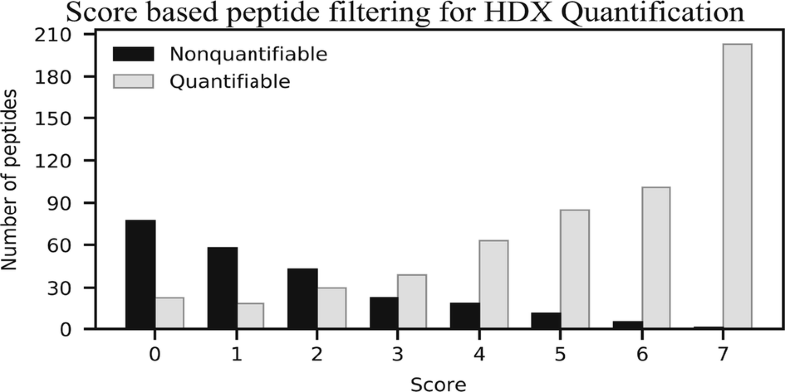
Hydrogen deuterium exchange measured by mass spectrometry (HDX-MS) is a commonly used technique for studying the structural dynamics of proteins in solution. The first part of any bottom-up HDX-MS experiment is to identify the peptides generated from a digestion step. This requires manual inspection of the identified peptides to determine their use for HDX-MS analysis, which is a time-consuming task. Throughout the literature, there have been different approaches for removing peptides that do not yield quantifiable HDX information. This includes using validity scores from the software used in the generation of the peptide map and that the peptide should be found in two out of three technical replicate experiments. Here, we analyze the previously available methods for filtering the identified peptides in regard to their ability to predict whether a peptide will provide quantifiable HDX-MS data or not. We also present a new score-based system relying on a combination of MS/MS parameters that offers an improved method for separating quantifiable peptides from the nonquantifiable. Using this score-based method reduces the number of peptide spectra that needs to be manually inspected and thereby the time spent curating HDX-MS data.
ᅟ
中文翻译:
优化的工作流程以选择用于HDX-MS数据分析的肽
通过质谱法(HDX-MS)测量的氢氘交换是研究溶液中蛋白质结构动力学的常用技术。自下而上的HDX-MS实验的第一部分是鉴定从消化步骤中产生的肽。这需要人工检查鉴定出的肽,以确定其在HDX-MS分析中的用途,这是一项耗时的任务。在整个文献中,已经有不同的方法来去除不能产生可量化的HDX信息的肽。这包括使用肽图生成中使用的软件的有效性评分,并且应在三个技术重复实验中的两个中找到该肽。这里,我们分析了先前可用的方法来过滤已识别的肽,以预测它们是否能够提供可量化的HDX-MS数据。我们还提出了一种新的基于分数的系统,该系统依赖于MS / MS参数的组合,提供了一种改进的方法,可从不可量化的肽中分离出可量化的肽。使用这种基于评分的方法减少了需要手动检查的肽谱数量,从而减少了整理HDX-MS数据所花费的时间。
ᅟ








































 京公网安备 11010802027423号
京公网安备 11010802027423号