当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Prediction and Analysis of the UV/Visible Absorption and Emission Spectra of Chiral Carbon Nanorings
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-08-30 00:00:00 , DOI: 10.1021/acs.jpca.8b07270 Rathawat Daengngern 1, 2 , Cristopher Camacho 2, 3 , Nawee Kungwan 4, 5 , Stephan Irle 2, 6
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-08-30 00:00:00 , DOI: 10.1021/acs.jpca.8b07270 Rathawat Daengngern 1, 2 , Cristopher Camacho 2, 3 , Nawee Kungwan 4, 5 , Stephan Irle 2, 6
Affiliation
|
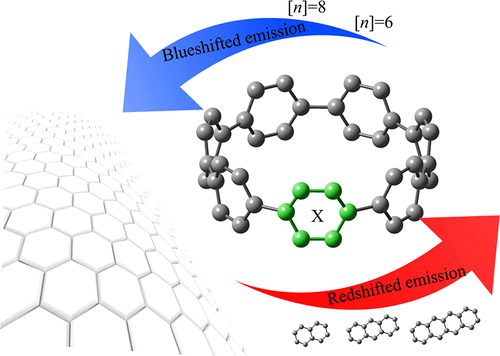
UV/vis absorption and emission spectra of recently synthesized chiral carbon nanorings were simulated using first-principles-based molecular dynamics and time-dependent density functional theory (TD-DFT). The chiral carbon nanorings are derivatives of the [n]cycloparaphenylene ([n]CPP) macrocycles, containing an acene unit such as naphthalene, ([n]CPPN), anthracene ([n]CPPA), and tetracene ([n]CPPT), in addition to n paraphenylene units. In order to study the effect of increasing molecular size on absorption and emission spectra, we investigated the cases where n = 6 and 8. Frontier molecular orbital analysis was carried out to give insight into the degree of excitation delocalization and its relationship to the predicted absorption spectra. The lowest excited singlet state S1 corresponds to a HOMO–LUMO π–π* transition, which is allowed in all chiral carbon nanorings due to lack of molecular symmetry, in contrast to the forbidden HOMO–LUMO transition in the symmetric [n]CPP molecules. The S1 absorption peak exhibits a blue-shift with increasing number of paraphenylene units in particular for [n]CPPN and [n]CPPA and less so in the case of [n]CPPT. In the case of CPPN and CPPA, the transition density is mainly localized over a semicircle of the macrocycle with the acene unit in its center but is strongly localized on the tetracene unit in the case of CPPT. Molecular dynamics simulations performed on the excited state potential energy surfaces reveal red-shifted emission of these chiral carbon nanorings when the size of the π-conjugated acene units is increased, although the characteristic [n]CPP blue-shift with increasing paraphenylene unit number n remains apparent. The anomalous emission blue-shift is caused by the excited state bending and torsional motions that stabilize the π HOMO and destabilize the π* LUMO, resulting in an increasing HOMO–LUMO gap.
中文翻译:

手性碳纳米环的紫外/可见吸收和发射光谱的理论预测和分析
使用基于第一原理的分子动力学和随时间变化的密度泛函理论(TD-DFT),模拟了最近合成的手性碳纳米环的UV / vis吸收和发射光谱。手性碳纳米环是[ n ]环对亚苯基([ n ] CPP)大环的衍生物,包含并苯单元,例如萘,([ n ] CPPN),蒽([ n ] CPPA)和并四苯([ n ] CPPT)。 ),以及n个对亚苯基单元。为了研究增加分子大小对吸收光谱和发射光谱的影响,我们研究了其中n= 6和8。进行了前沿分子轨道分析,以洞悉激发离域化程度及其与预测吸收光谱的关系。最低的激发单重态S 1对应于HOMO-LUMOπ-π*跃迁,与对称[ n ] CPP中禁止的HOMO-LUMO跃迁相反,由于缺乏分子对称性,所有手性碳纳米环中都允许该跃迁。分子。在S 1吸收峰显示出蓝移的增加的特别对苯单元的数量为[ Ñ ] CPPN和[ Ñ ] CPPA,而较少在[的情况下Ñ] CPPT。在CPPN和CPPA的情况下,过渡密度主要位于并苯中心位于大环的半圆上,而在CPPT的情况下,过渡密度主要位于并四苯单元上。尽管增加了对位亚苯基单元数n的特征[ n ] CPP蓝移,但当π共轭并苯单元的尺寸增加时,在激发态势能面上进行的分子动力学模拟显示这些手性碳纳米环的红移发射。仍然很明显。异常发射蓝移是由激发态弯曲和扭转运动引起的,它们使πHOMO稳定并使π* LUMO不稳定,从而导致HOMO-LUMO间隙增大。
更新日期:2018-08-30
中文翻译:
手性碳纳米环的紫外/可见吸收和发射光谱的理论预测和分析
使用基于第一原理的分子动力学和随时间变化的密度泛函理论(TD-DFT),模拟了最近合成的手性碳纳米环的UV / vis吸收和发射光谱。手性碳纳米环是[ n ]环对亚苯基([ n ] CPP)大环的衍生物,包含并苯单元,例如萘,([ n ] CPPN),蒽([ n ] CPPA)和并四苯([ n ] CPPT)。 ),以及n个对亚苯基单元。为了研究增加分子大小对吸收光谱和发射光谱的影响,我们研究了其中n= 6和8。进行了前沿分子轨道分析,以洞悉激发离域化程度及其与预测吸收光谱的关系。最低的激发单重态S 1对应于HOMO-LUMOπ-π*跃迁,与对称[ n ] CPP中禁止的HOMO-LUMO跃迁相反,由于缺乏分子对称性,所有手性碳纳米环中都允许该跃迁。分子。在S 1吸收峰显示出蓝移的增加的特别对苯单元的数量为[ Ñ ] CPPN和[ Ñ ] CPPA,而较少在[的情况下Ñ] CPPT。在CPPN和CPPA的情况下,过渡密度主要位于并苯中心位于大环的半圆上,而在CPPT的情况下,过渡密度主要位于并四苯单元上。尽管增加了对位亚苯基单元数n的特征[ n ] CPP蓝移,但当π共轭并苯单元的尺寸增加时,在激发态势能面上进行的分子动力学模拟显示这些手性碳纳米环的红移发射。仍然很明显。异常发射蓝移是由激发态弯曲和扭转运动引起的,它们使πHOMO稳定并使π* LUMO不稳定,从而导致HOMO-LUMO间隙增大。










































 京公网安备 11010802027423号
京公网安备 11010802027423号