当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Eliminating symmetry problems in electronegativity equalization and correcting self-interaction errors in conceptual DFT
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-08-24 , DOI: 10.1002/jcc.25356 László von Szentpály 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-08-24 , DOI: 10.1002/jcc.25356 László von Szentpály 1
Affiliation
|
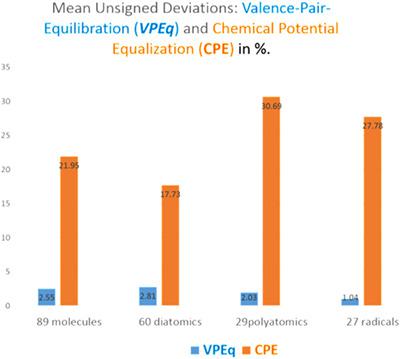
The chemical potential is by definition constant in molecules, and electronic charge is in principle equilibrated by bonding. Does electronegativity offer the best scale to unify these principles? According to conceptual density functional theory (c-DFT), the electronegativity equalization (ENE) and chemical potential equalization (CPE) principles seem rigorous and identical. However, the operational formulations of CPE and ENE fail to validate this claim, and frequently dramatic deviations from equalization are reported. We here eliminate the deviations to a very large extent. The problems originate from (i) c-DFT's exclusive reference to ground states and violations of the Wigner-Witmer symmetry constraints for bonding, (ii) electron self-interaction and delocalization errors. The problems are solved, and much more accurate ENE and bond polarities are obtained by replacing the ground-state electronegativity (χGS ) by the valence-state electronegativity (χVS ) and its generalization, the valence-pair-affinity (VPA, αVP ). The VPA is a charge dependent pair-sharing potential connected to Ruedenberg's bond theory that emphasizes the role of electron pair-density. The performances of the valence-pair equilibration (VPEq) and c-DFT's operational CPE are compared for 89 molecules with very diverse bond characters, including the "exotic" dimers Be2 , Mg2 , B2 , C2 , and Mn2 . The accuracy of VPEq is about 9 times better than that of operational CPE. Without requiring ad hoc calibrations, the VPEq bond polarities agree very well with results of state-of-the-art population analyses, and charges derived from vibrational spectra. A paradigm shift emphasizing valence states seems in order for c-DFT. Electronegativity and the chemical potential should be regarded as separate properties. Copyright © 2018 Wiley Periodicals, Inc.
中文翻译:

消除电负性均衡中的对称问题并纠正概念 DFT 中的自相互作用错误
根据定义,分子中的化学势是常数,而电荷原则上是通过键合来平衡的。电负性是否提供了统一这些原则的最佳尺度?根据概念密度泛函理论 (c-DFT),电负性均衡 (ENE) 和化学势均衡 (CPE) 原理似乎严格且相同。然而,CPE 和 ENE 的操作公式无法验证这一说法,并且经常报告与均衡的显着偏差。我们在这里在很大程度上消除了偏差。这些问题源于 (i) c-DFT 对基态的独家参考和对键合的 Wigner-Witmer 对称约束的违反,(ii) 电子自相互作用和离域误差。问题解决了,通过将基态电负性 (χGS ) 替换为价态电负性 (χVS ) 及其推广,价对亲和性 (VPA, αVP ),可以获得更准确的 ENE 和键极性。VPA 是一种依赖于电荷的对共享电位,与 Ruedenberg 的键理论相关,该理论强调电子对密度的作用。价对平衡 (VPEq) 的性能和 c-DFT 的操作 CPE 的性能比较了 89 个具有非常不同键特征的分子,包括“外来”二聚体 Be2、Mg2、B2、C2 和 Mn2。VPEq 的准确度比操作 CPE 的准确度高约 9 倍。无需特别校准,VPEq 键极性与最先进的种群分析结果和来自振动光谱的电荷非常吻合。强调价态的范式转变似乎是为了 c-DFT。电负性和化学势应被视为独立的特性。版权所有 © 2018 Wiley Periodicals, Inc.
更新日期:2018-08-24
中文翻译:
消除电负性均衡中的对称问题并纠正概念 DFT 中的自相互作用错误
根据定义,分子中的化学势是常数,而电荷原则上是通过键合来平衡的。电负性是否提供了统一这些原则的最佳尺度?根据概念密度泛函理论 (c-DFT),电负性均衡 (ENE) 和化学势均衡 (CPE) 原理似乎严格且相同。然而,CPE 和 ENE 的操作公式无法验证这一说法,并且经常报告与均衡的显着偏差。我们在这里在很大程度上消除了偏差。这些问题源于 (i) c-DFT 对基态的独家参考和对键合的 Wigner-Witmer 对称约束的违反,(ii) 电子自相互作用和离域误差。问题解决了,通过将基态电负性 (χGS ) 替换为价态电负性 (χVS ) 及其推广,价对亲和性 (VPA, αVP ),可以获得更准确的 ENE 和键极性。VPA 是一种依赖于电荷的对共享电位,与 Ruedenberg 的键理论相关,该理论强调电子对密度的作用。价对平衡 (VPEq) 的性能和 c-DFT 的操作 CPE 的性能比较了 89 个具有非常不同键特征的分子,包括“外来”二聚体 Be2、Mg2、B2、C2 和 Mn2。VPEq 的准确度比操作 CPE 的准确度高约 9 倍。无需特别校准,VPEq 键极性与最先进的种群分析结果和来自振动光谱的电荷非常吻合。强调价态的范式转变似乎是为了 c-DFT。电负性和化学势应被视为独立的特性。版权所有 © 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号