npj Computational Materials ( IF 9.4 ) Pub Date : 2018-08-24 , DOI: 10.1038/s41524-018-0103-x Hongxiang Zong , Ghanshyam Pilania , Xiangdong Ding , Graeme J. Ackland , Turab Lookman
|
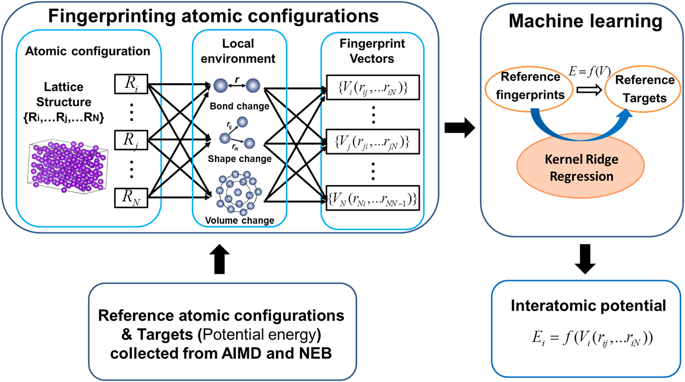
Atomic simulations provide an effective means to understand the underlying physics of structural phase transformations. However, this remains a challenge for certain allotropic metals due to the failure of classical interatomic potentials to represent the multitude of bonding. Based on machine-learning (ML) techniques, we develop a hybrid method in which interatomic potentials describing martensitic transformations can be learned with a high degree of fidelity from ab initio molecular dynamics simulations (AIMD). Using zirconium as a model system, for which an adequate semiempirical potential describing the phase transformation process is lacking, we demonstrate the feasibility and effectiveness of our approach. Specifically, the ML-AIMD interatomic potential correctly captures the energetics and structural transformation properties of zirconium as compared to experimental and density-functional data for phonons, elastic constants, as well as stacking fault energies. Molecular dynamics simulations successfully reproduce the transformation mechanisms and reasonably map out the pressure–temperature phase diagram of zirconium.
中文翻译:
通过机器学习开发锆马氏体相变的原子间势
原子模拟提供了一种有效的手段,以了解结构相变的基本物理原理。然而,由于经典的原子间电势不能代表多种键合,这对于某些同素异形金属仍然是一个挑战。基于机器学习(ML)技术,我们开发了一种混合方法,其中可以从头算分子动力学模拟(AIMD)高度保真地学习描述马氏体相变的原子间电势。使用锆作为模型系统,但缺乏描述相变过程的足够的半经验潜力,我们证明了该方法的可行性和有效性。具体来说,与声子,弹性常数以及堆积断层能的实验数据和密度函数数据相比,ML-AIMD原子间势能正确地捕获了锆的能量学和结构转变特性。分子动力学模拟成功地再现了转变机理,并合理地绘制了锆的压力-温度相图。










































 京公网安备 11010802027423号
京公网安备 11010802027423号