当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unusual Fluorine Substitution Effect on S···Cl Bonding between Sulfides and Atomic Chlorine
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-08-20 00:00:00 , DOI: 10.1021/acs.jpca.8b04495 Dipankar Sutradhar 1 , Sumitra Bhattarai 1 , Therese Zeegers-Huyskens 2 , Asit K. Chandra 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-08-20 00:00:00 , DOI: 10.1021/acs.jpca.8b04495 Dipankar Sutradhar 1 , Sumitra Bhattarai 1 , Therese Zeegers-Huyskens 2 , Asit K. Chandra 1
Affiliation
|
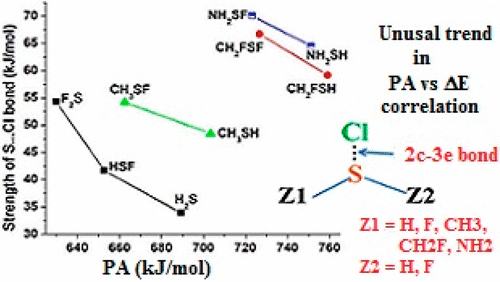
A theoretical investigation on the interaction of various sulfides and their fluorinated counterparts (H2S, HSF, F2S, CH3SH, CH3SF, CH2FSH, CH2FSF, NH2SH, NH2SF) with atomic chlorine has been carried out using density functional theory (DFT) based LC-BLYP/aug-cc-pVTZ and sophisticated ab initio CCSD(T)/aug-cc-pVQZ methods. The present study is intended to discuss the influence of the substituents implanted at the sulfur atom on the bonding parameters. The optimized geometries reveal that intermolecular S···Cl distances are short and range between 2.423 and 2.561 Å. A strong contraction of the S–F bond is also predicted. Two-center–three-electron S···Cl bonds are formed; the interaction energies are large and range from −33.9 to −70.1 kJ mol–1. Very surprisingly, the interaction energies are greater and the intermolecular distances are shorter for F-substituted sulfides than for unsubstituted ones. This is in complete contrast with the lower proton affinities and less negative electrostatic potentials of fluorinated sulfides. AIM analysis, the charge transfer from the sulfur atom to the Cl atom, and the spin densities on the Cl and S atoms are considered to explain this unusual behavior. The hyperconjugation energies from the LP(F) to the σ*(S–Cl) antibonding orbital can be considered as one of the stabilizing factors for the greater stability of the fluorinated complexes over the nonfluorinated ones.
中文翻译:

异常氟取代对硫化物与原子氯之间S··Cl键的影响
各种硫化物及其氟化物(H 2 S,HSF,F 2 S,CH 3 SH,CH 3 SF,CH 2 FSH,CH 2 FSF,NH 2 SH,NH 2 SF)与原子相互作用的理论研究氯已使用基于密度泛函理论(DFT)的LC-BLYP / aug-cc-pVTZ和复杂的从头算进行CCSD(T)/ aug-cc-pVQZ方法。本研究旨在讨论在硫原子上注入的取代基对键合参数的影响。优化的几何结构表明分子间S··Cl距离很短,范围在2.423至2.561Å之间。还可以预测到SF键的强烈收缩。形成两个中心的三电子S···Cl键;相互作用能很大,范围为−33.9至−70.1 kJ mol –1。非常令人惊讶的是,与未取代的硫化物相比,F取代的硫化物的相互作用能更大,分子间距离更短。这与较低的质子亲合力和较低的氟化硫化物负静电势完全相反。AIM分析,电荷从硫原子转移到Cl原子以及Cl和S原子上的自旋密度被认为是解释这种异常行为的原因。从LP(F)到σ*(S–Cl)反键轨道的超共轭能被认为是稳定的因素之一,因为氟化物比非氟化物具有更高的稳定性。
更新日期:2018-08-20
中文翻译:
异常氟取代对硫化物与原子氯之间S··Cl键的影响
各种硫化物及其氟化物(H 2 S,HSF,F 2 S,CH 3 SH,CH 3 SF,CH 2 FSH,CH 2 FSF,NH 2 SH,NH 2 SF)与原子相互作用的理论研究氯已使用基于密度泛函理论(DFT)的LC-BLYP / aug-cc-pVTZ和复杂的从头算进行CCSD(T)/ aug-cc-pVQZ方法。本研究旨在讨论在硫原子上注入的取代基对键合参数的影响。优化的几何结构表明分子间S··Cl距离很短,范围在2.423至2.561Å之间。还可以预测到SF键的强烈收缩。形成两个中心的三电子S···Cl键;相互作用能很大,范围为−33.9至−70.1 kJ mol –1。非常令人惊讶的是,与未取代的硫化物相比,F取代的硫化物的相互作用能更大,分子间距离更短。这与较低的质子亲合力和较低的氟化硫化物负静电势完全相反。AIM分析,电荷从硫原子转移到Cl原子以及Cl和S原子上的自旋密度被认为是解释这种异常行为的原因。从LP(F)到σ*(S–Cl)反键轨道的超共轭能被认为是稳定的因素之一,因为氟化物比非氟化物具有更高的稳定性。








































 京公网安备 11010802027423号
京公网安备 11010802027423号